Tandem mass spectrometry provides more than a list of fragment ion masses. The fragmentation pattern is the joint outcome of precursor chemistry, activation method, collision energy regime, charge state, adduct form, instrument architecture, and sample complexity. Interpreting this pattern — and selecting the right fragmentation strategy for a given analytical question — requires understanding not just what fragments are produced, but why they are produced and what level of evidence each fragment type supports. This guide is for researchers who already understand what CID, HCD, ETD, and other fragmentation methods are. It focuses on translating that knowledge into method selection, spectral validation, and evidence-level decisions for RUO projects — covering collision energy optimization, transition selection, PTM localization, unknown metabolite annotation, lipid interpretation, glycopeptide characterization, and pre-submission project communication. For further reading on this topic, see our dedicated resource on lc-ms vs lc-ms/ms.
Customized LC-MS/MS method development services support project-specific fragmentation strategy design based on analyte type, evidence requirements, and sample complexity.
Figure 1: Current page vs new page content boundary — fragmentation physics (existing) vs method decision and evidence ranking (this guide)
This Is Not Another Ion Type Glossary
The existing resource on mass spectrometry ion types and fragmentation patterns already covers protonated, sodiated, and deprotonated ions, isotope envelope and formula filtering, even-electron vs odd-electron ions, the mobile proton model, CID/HCD/ETD/ECD/UVPD mechanisms, neutral loss and diagnostic ions, collision energy effects on pathway competition, chimeric spectra, and database search vs de novo interpretation. This guide does not repeat that content. It starts from the premise that fragmentation pattern is not a peak list — it is a joint product of method selection, evidence level, and structural conclusion boundaries.
MOFU/BOFU clients who have read the mechanism-level content now ask different questions: Should my target molecule be analyzed by CID, HCD, ETD, EThcD, UVPD, or MS3? Why does the same precursor produce different spectra on different platforms? Does a high search score mean the structural evidence is sufficient? Do I need routine identification, site localization, structural elucidation, or quantitative transition development? This guide answers those questions directly.
Start from the Analytical Claim, Not the Fragmentation Mode
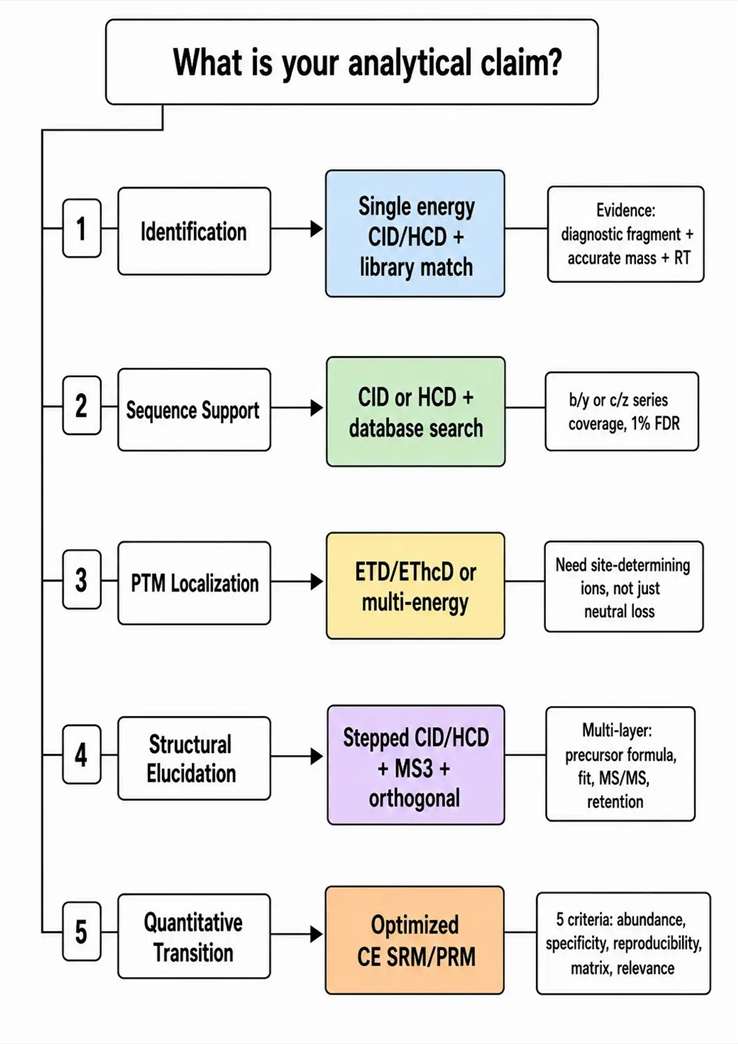
The most common client request — "Can you perform MS/MS fragmentation analysis?" — is too broad to inform method design. Different conclusions require different levels of spectral evidence, and fragmentation strategy should be selected by the analytical claim, not by method familiarity.
Five types of analytical claims and their evidence requirements:
Identification — confirming the presence of a known compound class. Requires accurate mass and isotopic pattern, retention time, and at least one diagnostic fragment or library match. Fragmentation: single-energy CID or HCD with library matching.
Sequence support — assigning peptide or protein identity. Requires b/y or c/z ion series coverage, typically 1% FDR at the peptide level. Fragmentation: CID or HCD for routine bottom-up; ETD for highly charged or modified precursors.
PTM localization — determining the specific residue carrying a modification. Requires site-determining ions that differentiate modified from unmodified forms of the same peptide. Fragmentation: ETD/EThcD or multi-energy acquisition to preserve labile modifications while generating backbone cleavage.
Structural elucidation — resolving the complete structure of an unknown compound, including isomeric and regiochemical features. Requires multiple orthogonal evidence layers: precursor formula, isotope fit, multi-energy MS/MS, adduct comparison, retention behavior, and often MSn or standard comparison. Fragmentation: stepped CID/HCD with follow-up targeted MS2 or MS3.
Quantitative transition design — selecting product ions for MRM or PRM assays. Requires abundance, specificity, reproducibility, matrix robustness, and structural relevance — not just the most intense peak in the MS2 spectrum. Fragmentation: CID or HCD at optimized CE with qualification against blank matrix extracts.
Common interpretation errors to avoid: Treating a single diagnostic ion as complete structural confirmation. Treating a high database search score as sufficient PTM site localization. Treating a single-collision-energy spectrum as the complete fragmentation behavior of the molecule under all conditions. Each of these errors leads to the wrong fragmentation strategy being selected for the next experiment.
High-resolution exact-mass confirmation workflows provide the mass accuracy foundation required before fragmentation strategy decisions. Unknown-metabolite identification workflows apply multi-evidence structural elucidation strategies.
Figure 2: Analytical claim to fragmentation strategy decision tree — define the evidence goal first, then select the method
Routine Identification vs Structural Elucidation: Two Different Evidence Games
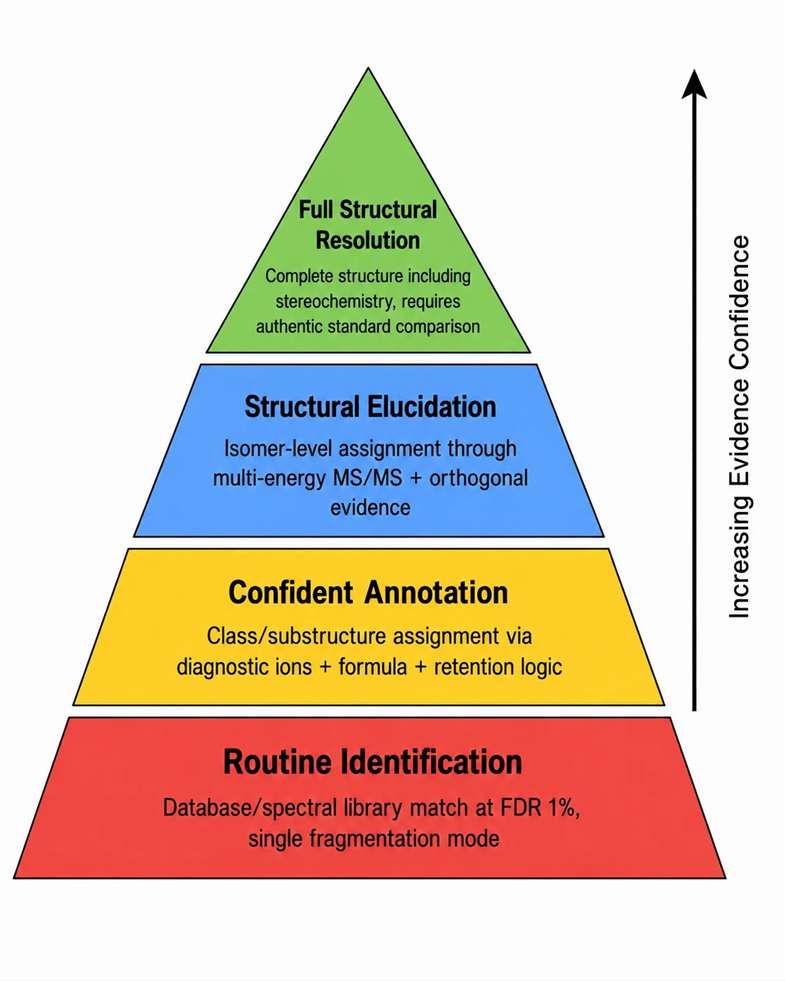
Routine identification and structural elucidation operate at fundamentally different evidence levels, and the fragmentation strategy that supports one may be insufficient for the other.
Routine identification is appropriate for: peptide and protein identification by database search, known metabolite matching against spectral libraries, known lipid class assignment by diagnostic ions, and standard compound confirmation by retention time and MS2 match. These applications rely on reference data — libraries, databases, or standards — and the fragmentation strategy is optimized for compatibility with reference conditions.
Structural elucidation is required for: unknown metabolite annotation where no standard or library entry exists, novel impurity identification in pharmaceutical development, modified peptide analysis where the modification is unexpected, glycopeptide or lipid structural interpretation beyond class-level assignment, and unexpected fragment family investigation where the fragmentation behavior does not match any reference. These applications require multi-layer evidence because no single reference point exists.
Key distinction: Routine identification can succeed with a single fragmentation method at standard conditions. Structural elucidation requires multiple collision energies, complementary activation methods, retention behavior analysis, adduct comparison, and often orthogonal LC conditions. The evidence threshold is fundamentally higher. A confident routine identification at FDR 1% may provide class-level information only for the same data in an elucidation context — this distinction is critical when setting project expectations with clients.
Protein-level identification in complex discovery datasets follows routine database search workflows. De novo sequencing strategies address cases where database matching is insufficient due to incomplete fragment ladders or unexpected modifications.
Figure 3: MS/MS evidence pyramid — evidence thresholds from "may be present" through "full structural resolution"
Choosing CID, HCD, ETD/EThcD, UVPD, or MS3 by Question Type
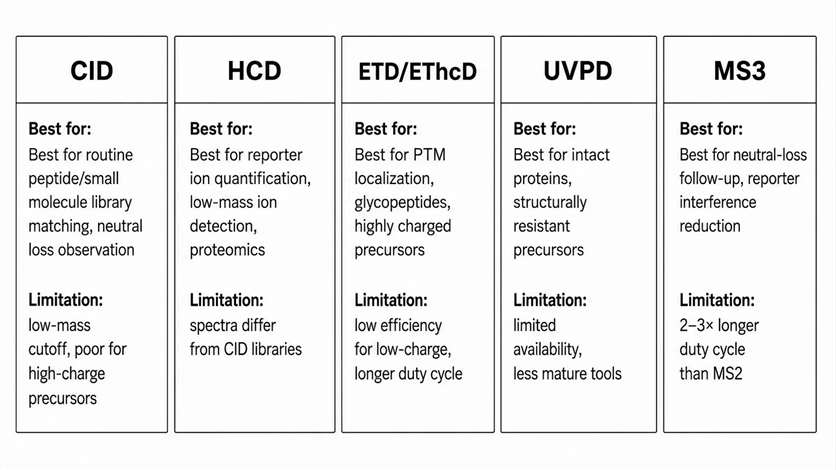
Each activation method deposits energy into the precursor ion through a different mechanism, producing characteristic fragment types and coverage patterns. The choice between them should be guided by the analytical question, not by instrument availability.
CID uses multiple low-energy collisions with neutral gas. Best for: routine peptide fragmentation producing b/y ion series, small molecule product ion screening where library matching to NIST or mzCloud is the goal, neutral-loss behavior observation for labile functional groups, and ion trap MSn exploration where sequential fragmentation events are needed. Limitation: low-mass fragment cutoff below approximately 1/3 of precursor m/z in ion trap CID, and poor fragmentation of highly charged or bulky precursors.
HCD uses higher-energy collisions in a dedicated cell. Best for: reporter ion quantification (TMT, iTRAQ) where low-mass reporter detection is essential, higher-energy peptide fragmentation producing additional immonium and internal ions, low-mass ion observation that CID ion traps cannot access, and many standard discovery proteomics workflows. Limitation: HCD spectra may differ from CID library entries for the same compound, requiring platform-specific library matching.
ETD/ECD uses electron transfer/dissociation to fragment highly charged precursors. Best for: highly charged peptide and protein ions (charge state ≥3+), labile PTM retention where collisional methods would strip the modification, top-down or middle-down characterization of intact proteins, and modification-rich analytes where backbone coverage must be preserved. Limitation: low fragmentation efficiency for low-charge precursors; requires longer ion-ion reaction times that increase duty cycle.
EThcD combines ETD with supplemental HCD activation. Best for: modified peptides and glycopeptides requiring both c/z ions (from ETD) and b/y ions (from HCD) for complete sequence and site assignment. Ideal for cases where ETD alone produces insufficient fragment ion yield. Limitation: higher data complexity requiring specialized search algorithms.
UVPD uses ultraviolet photons for activation. Best for: intact protein fragmentation producing a broad range of ion types (a, b, c, x, y, z), structurally resistant precursors that do not fragment well by CID or ETD, and selected advanced characterization scenarios where extended fragment coverage is needed. Limitation: currently limited instrument availability and less mature data analysis tools.
MS3 fragments a selected product ion from a primary MS2 event. Best for: neutral-loss-dominated precursors where the primary fragment is uninformative, reporter-ion interference reduction in TMT quantification, ambiguous product ion origin investigation, and structural pathway confirmation for unknown compounds. Limitation: 2-3× longer duty cycle than MS2 alone, reducing the number of precursors that can be interrogated per run.
MS platform-based fragmentation strategy selection ensures compatibility between analytical goals and available instrumentation. Top-down PTM characterization strategies apply ETD and UVPD for intact protein analysis. Glycopeptide characterization workflows combine HCD and EThcD for complementary evidence.
Figure 4: Fragmentation method selection matrix — CID, HCD, ETD/EThcD, UVPD, and MS3 matched to analytical goals
Collision Energy Optimization Is Pathway Selection, Not Intensity Tuning
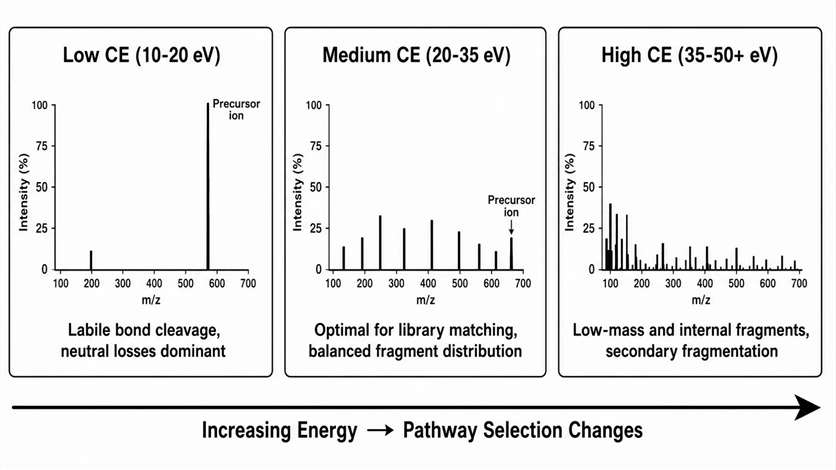
Collision energy does not simply make a spectrum "more intense" or "less intense." It selects which fragmentation pathways are energetically accessible, changing the distribution of fragment ion types and their relative abundances. The collision energy is a method design parameter that must be matched to the analytical goal.
Low energy regime (10-20 eV for typical small molecules, 15-25% NCE for peptides): The precursor ion remains dominant. Only the most labile bonds fragment, producing neutral losses (water, ammonia, carbon dioxide) and early-cleavage pathway products. Useful for observing precursor stability, confirming the presence of labile functional groups, and generating initial fragmentation pathways for unknown compounds.
Medium energy regime (20-35 eV / 25-35% NCE): The precursor declines to 10-30% relative abundance. A balanced set of sequence ions or product ions emerges, representing the optimal condition for library matching and routine identification. Most reference spectra in NIST and mzCloud are acquired in this regime.
High energy regime (35-50+ eV / 35-50% NCE): The precursor is fully depleted. Low-mass ions, immonium ions, internal fragments, and secondary fragments become dominant. Useful for obtaining diagnostic low-mass information and for generating additional sequence coverage in proteomics, but higher risk of producing non-specific fragments that reduce search specificity.
Method development logic: A stepped collision energy approach — acquiring MS2 spectra at 3-5 energies across the low-to-high range — provides the most complete fragmentation information for structural elucidation. For routine library matching, a single medium-energy spectrum is sufficient. For quantitative MRM/PRM transition development, the optimal CE is defined by the transition signal-to-noise ratio, not by the MS2 spectral quality — and must be validated in the target matrix, not in neat solution. A transition that is intense in a standard solution but suppressed in plasma is not a useful transition.
MRM transition development applies CE optimization specific to each target analyte and matrix combination. PRM assay design uses full product-ion confirmation at optimized collision energy.
Figure 5: Collision energy ramp and fragmentation pathway distribution — CE selects pathway, not intensity
Transition Selection for MRM/PRM: The Fragment Ion Must Be Useful, Not Just Abundant
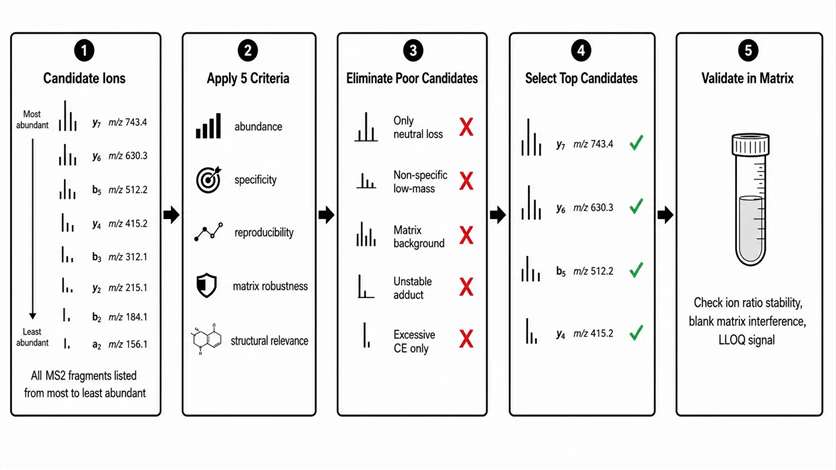
Selecting a product ion for quantitative transition monitoring requires five criteria, only one of which is abundance.
Abundance: The transition must provide sufficient signal at the required LLOQ. Without adequate abundance, the quantitative range is compromised.
Specificity: The transition must distinguish the target analyte from potential interferences in the sample matrix. A transition may look clean in a neat standard solution but suffer from interference in plasma or tissue homogenate.
Reproducibility: The transition must produce stable peak area ratios across injections, days, and sample batches. Ion ratio variability >20% indicates an unstable transition, often due to competing fragmentation pathways at the selected CE.
Matrix robustness: The transition must not be suppressed or enhanced by co-eluting matrix components. Post-column infusion experiments identify retention time windows where matrix effects are strongest.
Structural relevance: The transition should be chemically reasonable for the target structure. A non-specific low-mass ion, a fragment from an unstable adduct, or a fragment only present at excessive collision energy may pass the first four criteria but fail structural logic, leading to false positives in method validation.
Transitions to avoid: Only neutral-loss ions (e.g., loss of water or ammonia only, no backbone fragments), non-specific low-mass ions common to many compounds, matrix-rich background ions that overlap with endogenous compounds, fragments from unstable adducts that may not form reproducibly, and fragments only present at collision energies above 50 eV where secondary fragmentation compromises specificity.
SRM/MRM quantitative transition strategy applies the five-criteria workflow. Chromatographic separation support improves MS/MS specificity by resolving interferences before the mass spectrometer.
Figure 6: Quantifier and qualifier transition selection workflow — from candidate product ions to validated assay
When a High Search Score Is Not Enough
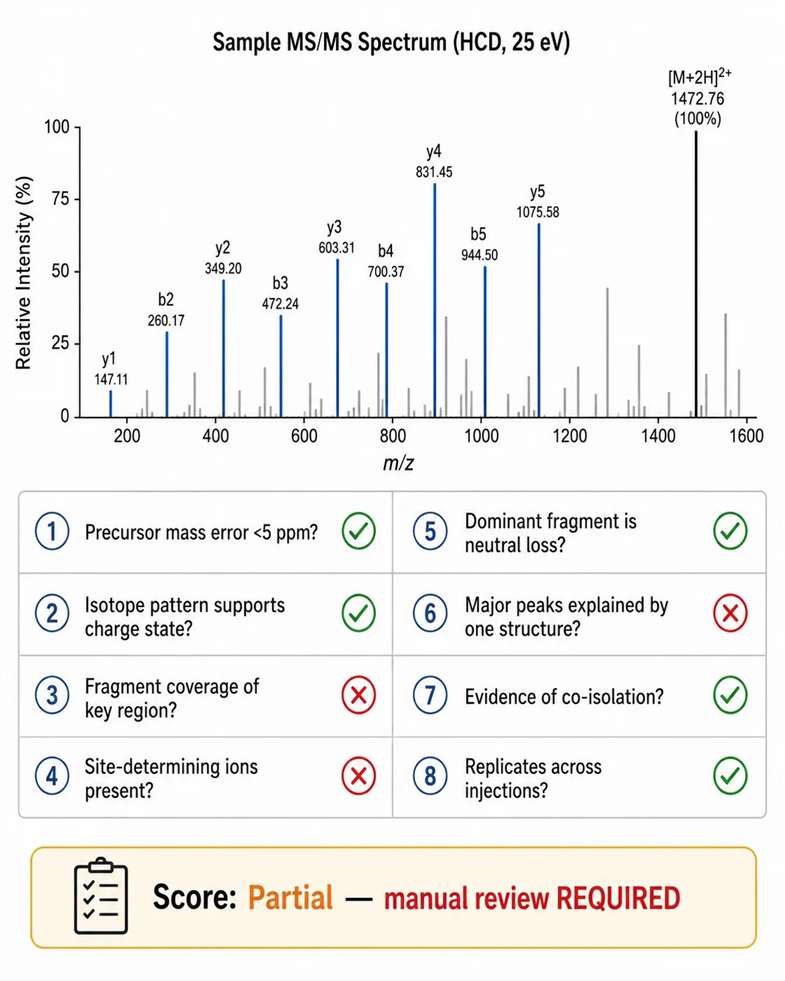
Database search algorithms assign statistical confidence scores to peptide-spectrum matches (PSMs), but a high score does not guarantee that the structural evidence is complete or correct. Several scenarios produce high scores with incomplete evidence.
Chimeric spectra: When two or more precursors are co-isolated, the composite MS2 spectrum may match one of them with high score while the second precursor's fragments are present but unassigned. Manual inspection reveals peaks that do not belong to the assigned precursor — a signature of chimeric interpretation.
Missing site-determining ions: For PTM localization, a high PSM score may be driven by non-localizing backbone fragments while the site-determining ions that differentiate modified from unmodified residues are absent. The score reflects peptide identification, not localization confidence.
Dominant neutral loss: For labile modifications such as phosphorylation or sulfation, the most abundant fragment is often a neutral loss, not a sequence-determining ion. A spectrum dominated by neutral loss can achieve a high match score against the unmodified peptide, suggesting the modification is present without localizing it.
Manual spectrum review checklist for high-scoring but suspicious matches: Is the precursor mass error within 5 ppm? Does the isotope pattern support the assigned charge state and formula? Do fragment ions cover the key regions of the sequence? Are site-determining ions present for PTM assignments? Is the dominant fragment explained by neutral loss rather than sequence cleavage? Can the major peaks be explained by a single consistent structure? Is there evidence of co-isolated precursor contributions? Each check that fails reduces the evidence level of the assignment, regardless of the search score.
PTM-focused downstream interpretation support provides automated and manual review of site-localizing evidence. De novo sequencing strategies address cases where search scores are high but unexpected fragments cannot be explained by the top database match.
Figure 7: High-score spectrum manual review checklist — what a search score does and does not guarantee
PTM Localization: Fragmentation Strategy Should Preserve the Modification and Localize It
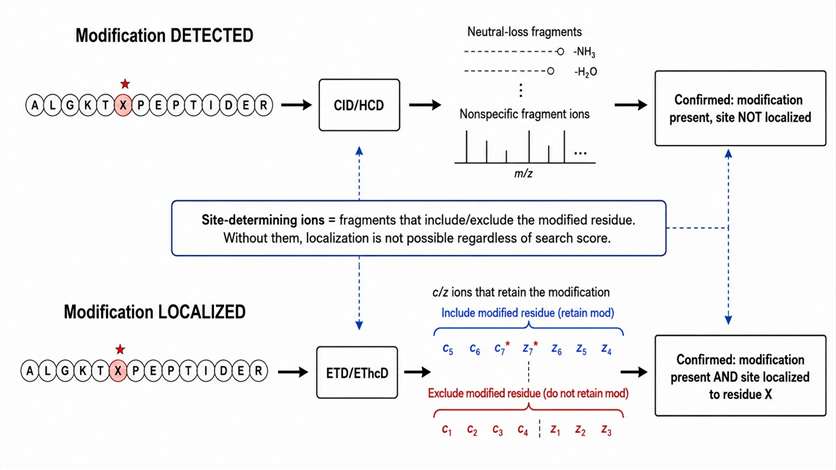
PTM analysis has two distinct goals: identifying the modified peptide and localizing the modification site. A fragmentation strategy that achieves the first goal may fail at the second.
Phosphorylation: CID and HCD produce characteristic neutral loss of phosphoric acid (98 Da for singly phosphorylated, 49 Da per charge for multiply charged). This neutral loss is evidence that the modification is present, but it is not site-determining. Site-determining ions are those that retain the phosphate group on a specific residue — if the peptide has multiple serine, threonine, or tyrosine residues, only fragments that include or exclude the modified residue can localize the site. ETD/EThcD preserves the phosphate during backbone cleavage, making it the preferred method when site localization is required.
Glycosylation: HCD produces strong glycan oxonium ions (m/z 204.087 for HexNAc, m/z 366.140 for HexNAcHex, etc.) that confirm the presence of a glycan but do not identify the glycosite. ETD/EThcD generates c/z ions that retain the glycan on the modified residue, enabling site assignment. A complete glycopeptide assignment requires both HCD-derived glycan composition and EThcD-derived peptide sequence and glycosite localization.
Acetylation and methylation: Mass shifts of +42.011 Da (acetylation) and +14.016 Da (methylation) must be localized to specific lysine or arginine residues. The fragment ions that bracket the modified residue distinguish between alternative modification sites. MS3 triggered by neutral loss can resolve ambiguous site assignments in collisional fragmentation data.
Phosphopeptide MS/MS site localization applies ETD/EThcD for confident site assignment. Top-down PTM characterization strategies preserve intact modification patterns on whole proteins.
Figure 8: PTM localization evidence pathway — distinguishing modification detection from site determination
Small Molecules and Metabolites: Diagnostic Ions Are Clues, Not Full Structures
For unknown small molecule identification, a single diagnostic ion provides class-level or substructure-level information but does not constitute complete structural confirmation. The evidence hierarchy for unknown metabolite annotation follows defined levels. For additional insights into physics metabolomics principles, explore our in-depth resource.
Class-level annotation is supported by a single diagnostic ion characteristic of a compound class — for example, m/z 184 for phosphocholine in lipidomics, or m/z 274 for glucuronide conjugates in metabolomics. The presence of this ion indicates that the compound belongs to the class but provides no information about the remainder of the structure.
Substructure annotation is supported by multiple related fragments that define a partial structure — for example, the presence of both the parent ion and a specific neutral loss that together define a substructure fragment. This level identifies a portion of the molecule but cannot resolve the complete structure without additional evidence.
Candidate ranking requires exact mass (<5 ppm), isotopic pattern fit, MS/MS consistency with each candidate, and retention time logic or prediction. Candidates that pass all four filters are ranked, but isomer-level discrimination requires additional MS2 evidence or standard comparison.
Confident confirmation requires matching to an authentic standard analyzed under identical LC-MS/MS conditions, meeting Level 1 identification criteria under the Metabolomics Standards Initiative framework.
LC-MS/MS untargeted metabolite profiling generates multi-energy MS/MS data for candidate ranking. Accurate mass and isotope-fit confirmation provides the precursor-level evidence required before fragment interpretation.
Charge State and Adduct Effects on Fragmentation Behavior
Precursor charge state and adduct form are not sample preparation details — they are primary determinants of fragmentation behavior that must be considered in method design.
Charge state effects: Higher charge states distribute available internal energy across more degrees of freedom, requiring higher collision energy to achieve equivalent fragmentation. A 2+ peptide fragments optimally at 25-30% NCE, while a 4+ peptide of the same sequence may require 30-35% NCE. ETD efficiency increases with charge state — triply charged precursors fragment readily by ETD while doubly charged precursors may not. This charge-state dependence means that switching between ESI and nanoESI (which produces different charge state distributions) can require re-optimization of the fragmentation method.
Adduct effects: Sodium adducts [M+Na]⁺ produce fundamentally different MS2 spectra than protonated [M+H]⁺ for the same molecule. The sodium ion localizes charge differently, redistributing fragmentation pathways and often stabilizing the precursor against dissociation. A library spectrum acquired from [M+H]⁺ will not match data from [M+Na]⁺. For quantitative analysis, [M+H]⁺ is preferred for transition reproducibility. For structural elucidation, both adduct forms provide complementary information: [M+H]⁺ spectra reveal labile bond cleavage, while [M+Na]⁺ spectra may reveal fragmentation pathways that are suppressed in the protonated form.
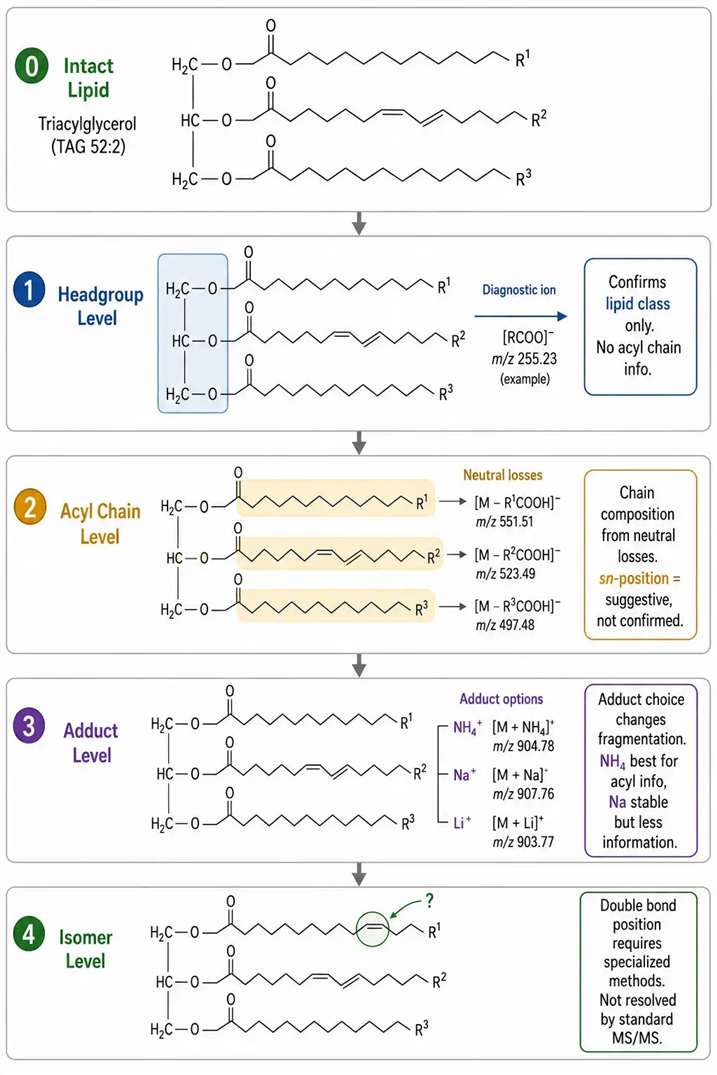
Lipid MS/MS: Headgroup, Acyl Chain, and Adduct Chemistry Must Be Read Separately
Lipid MS/MS interpretation differs fundamentally from peptide MS/MS. Peptide fragmentation produces a backbone ion ladder that directly encodes the sequence. Lipid fragmentation produces headgroup diagnostic ions, acyl chain neutral losses, and adduct-dependent cleavage patterns that must be interpreted as separate evidence layers.
Headgroup diagnostics: Phosphocholine (PC and SM) produces the characteristic m/z 184 ion in positive mode, confirming the headgroup class. This ion does not provide any information about the acyl chain composition, chain length, double bond position, or sn-position. For PE, PS, PI, and PA classes, negative mode fragmentation is often more informative — producing headgroup-specific carboxylate anions and acyl chain fragments that the positive mode may suppress.
Acyl chain evidence: Neutral loss of fatty acids from the sn-1 and sn-2 positions provides evidence for acyl chain composition. Relative loss abundances can suggest but not independently confirm sn-position assignment. Fragmentation in negative mode — producing fatty acid carboxylate anions — provides more reliable acyl chain evidence than positive mode for most glycerophospholipid classes. The two fatty acyl chains are distinguished by their specific neutral losses from the intact precursor ion.
Adduct effects on lipid fragmentation: The choice of adduct former in the mobile phase (ammonium acetate, sodium acetate, lithium acetate) fundamentally changes the fragmentation behavior of neutral lipids such as triacylglycerols and cholesterol esters. Ammonium adducts [M+NH₄]⁺ fragment to produce diacylglycerol-like product ions that reveal the fatty acyl composition. Sodium adducts are more stable and produce simpler spectra with less acyl chain information. Lithium adducts can enhance specific fragmentation pathways for double-bond position analysis, though this requires specialized method development.
Common lipid MS/MS interpretation errors: Using m/z 184 to claim complete PC structure identification — this ion confirms headgroup class only. Treating lipid sum composition as complete acyl-chain assignment — C36:2 does not specify which two fatty acids (18:0/18:2 vs 16:0/20:2). Ignoring adduct form effects on lipid annotation — the same lipid in different adduct forms produces different fragmentation patterns and may generate different database matches.
Targeted lipid readouts for class-resolved structural interpretation apply adduct-specific fragmentation methods.
Figure 9: Lipid MS/MS evidence layers — headgroup, acyl chain, adduct, and isomer-level interpretation boundaries
Glycopeptides and Glycans: Why One Fragmentation Mode Rarely Answers Everything
Glycopeptide MS/MS has a tripartite evidence requirement that no single fragmentation method can fully satisfy: glycan oxonium ions confirm that a glycan is present, peptide backbone ions identify the peptide sequence, and glycosite-localizing ions determine which residue carries the glycan.
HCD contributions: HCD produces strong glycan oxonium ions (m/z 204.087 for HexNAc, m/z 366.140 for HexNAcHex) that are diagnostic for glycosylation. HCD also generates peptide backbone b/y ions that support identification of the peptide portion. However, the glycan is typically lost during HCD fragmentation, meaning that glycosite-localizing evidence — the intact glycopeptide fragment containing both peptide and glycan — is missing from HCD data alone.
ETD contributions: ETD preserves the intact glycan on the peptide backbone during fragmentation, producing c/z ions that retain the modification. This enables glycosite localization by identifying which fragment contains the mass shift. The limitation is lower fragmentation efficiency for glycopeptides compared to HCD, particularly for low-charge or large glycopeptide precursors.
EThcD — the complementary solution: EThcD combines ETD with supplemental HCD activation, producing both c/z ions (with intact glycan for site localization) and b/y ions (for peptide sequence). For most glycopeptide characterization projects, EThcD provides the most complete evidence in a single acquisition. The trade-off is increased spectral complexity requiring specialized search algorithms that handle mixed fragmentation types.
Interpretation boundary: Strong glycan oxonium ions in an HCD spectrum confirm that a glycopeptide was fragmented, but they do not identify the specific peptide sequence, the glycosite, or the complete glycan structure. A complete glycopeptide assignment requires evidence from at least two complementary fragmentation modes.
Glycopeptide characterization workflows apply HCD and EThcD for complementary evidence.
Chimeric Spectra and Co-Isolation: The Silent Failure Mode in Complex Samples
In complex sample analysis, a spectrum that partially matches the expected fragmentation pattern may be more dangerous than a complete mismatch — it can pass automated quality filters while containing unassigned fragments from co-isolated precursors that lead to incorrect structural conclusions.
Common sources: Precursor co-isolation — two or more ions within the isolation window are fragmented together. Isotope overlap — the isotope peak of a high-abundance precursor falls within the isolation window of a lower-abundance target. Adduct overlap — a sodium adduct or other adduct of one compound falls at the same nominal m/z as a different compound. In-source fragments — fragments generated in the ion source from a different compound are isolated as independent precursors. Co-eluting isomers — structurally related compounds that co-elute and share similar precursor masses.
Detection and mitigation: Narrowing the isolation window from 2.0 Da to 0.4-0.7 Da reduces co-isolation risk but reduces precursor transmission. High-resolution MS2 helps resolve fragments from different precursors by accurate mass but does not eliminate chimeric interpretation risk. PRM acquisition with full MS2 spectra provides more data for chimeric spectrum identification than SRM with single transitions. Comparing fragment ion elution profiles across the chromatographic peak — fragments from the same precursor share identical elution profiles — is the most definitive test for chimeric spectra.
When to suspect chimeric data: Search score is high but significant fragments remain unexplained. Isotope pattern is inconsistent with assigned charge state. Key expected fragments are absent. High-scoring identification does not replicate across injections or collision energies.
Platform-to-Platform Differences: Why Spectral Libraries Do Not Always Transfer Cleanly
The same compound analyzed on different MS platforms often produces MS/MS spectra with different fragment relative abundances and, in some cases, different fragment identities. Understanding these differences is essential for cross-platform method transfer and library-based identification confidence.
Key variables affecting spectral comparability: Ion trap CID vs beam-type HCD produce different fragment distributions because ion trap CID has a low-mass cutoff near 1/3 of precursor m/z, while HCD detects the full fragment range. Normalized collision energy scales differ between manufacturers — 30% NCE on one platform is not equivalent to 30% on another. Collision cell design affects ion residence time and collision frequency. Scan speed and AGC settings affect ion statistics — faster scanning reduces signal-to-noise, potentially missing fragments critical for structure assignment.
Practical guidance: For library-based identification, use platform-specific spectral libraries when possible. For method transfer, confirm that key diagnostic fragments — not just the most abundant ones — are present with qualitatively similar abundance patterns. For quantitative transition transfer, validate specificity on the target platform against matrix blanks, as interference patterns differ between platforms.
Before submitting an inquiry for MS/MS fragmentation analysis, defining the following parameters improves project design efficiency and reduces scope ambiguity.
Information to prepare: Analyte type (peptide, protein, metabolite, lipid, glycan, glycopeptide). Analysis goal (identification, localization, structural elucidation, quantification). Sample matrix (cell, tissue, serum, plant, microbial, purified compound). Expected modification or compound class if known. Available reference standard (yes/no, concentration). Preferred ion mode if known. Previous MS1 or MS2 data if available. Required interpretation level.
Low-quality request patterns: "Need MS/MS analysis" (method scope undefined). "Need fragmentation pattern" (evidence level unspecified). "Need structure confirmation" (confirmation against what reference?). These requests require clarification before method design can begin.
High-quality request patterns: "Need to distinguish class-level from species-level lipid annotation." "Need PTM site localization, not only modified peptide identification." "Need MRM transition candidates for targeted quantification." "Need unknown metabolite candidate ranking with MS/MS evidence across multiple collision energies." These requests enable direct translation into a specific fragmentation strategy and evidence-level target.
Customized RUO MS/MS project design supports project scoping from initial inquiry through data delivery. Discuss RUO tandem MS requirements with scientific staff for method development planning.
FAQ
How do I choose between CID, HCD, ETD, and EThcD?
CID for routine peptide and small molecule library matching. HCD for reporter-ion quantification and low-mass fragment detection. ETD/EThcD for PTM localization and glycopeptide characterization. The choice should be guided by the analytical claim, not by instrument availability.
Is a diagnostic ion enough for compound identification?
A single diagnostic ion supports class-level annotation only. Confident structural identification requires multiple orthogonal evidence layers including exact mass, isotope fit, multi-energy MS/MS, and retention behavior, with standard comparison for Level 1 identification.
Why does collision energy change the MS/MS spectrum?
CE controls which fragmentation pathways are energetically accessible — low CE favors labile bond cleavage, high CE favors multiple bond cleavage and secondary fragmentation. The spectrum at a single CE represents one sampling of the molecule's fragmentation landscape, not the complete picture.
How are MRM transitions selected from product ions?
Five criteria: abundance, specificity, reproducibility, matrix robustness, and structural relevance. The most intense peak in the MS2 spectrum is not always the best transition — validate against blank matrix extracts before finalizing.
When is MS3 useful in tandem mass spectrometry?
MS3 is useful when the primary MS2 fragment is a neutral loss rather than a structure-informative ion, when TMT reporter-ion interference needs to be reduced, or when ambiguous product ion origin needs to be resolved by fragmenting a selected product ion.
Why can a high search score still require manual spectrum review?
Search scores reflect statistical match probability against reference data, not structural completeness. High scores can result from chimeric spectra, dominant but non-localizing neutral losses, or matches driven by a subset of peaks while critical evidence ions are absent.
References
- Integration of alternative fragmentation techniques into standard proteomics pipelines. Nature Methods. 2026;23:03042.
- Gonzalez de Vega R, et al. Dual-platform mass spectrometry for fragmentation pattern analysis in metabolomics. Analytical Chemistry. 2024;96:8221-8229.
- Optimal dissociation methods differ for N- and O-glycopeptide analysis. Nature Communications. 2020;11:5110.
- Lou R, et al. The hidden impact of in-source fragmentation in metabolic MS data interpretation. Nature Metabolism. 2024;6:1018-1031.
- Ivanisevic J, et al. From samples to insights into metabolism: LC-MS-based metabolomics guide. Nature Protocols. 2024;19:2093-2120.











