Meta intent: A mechanism-first, research-oriented article explaining how nucleotides function simultaneously as energetic intermediates, metabolic flux variables, and regulatory signals, with emphasis on pathway control, dNTP balance, and modern analytical strategy.
Nucleotides are often introduced as the building blocks of DNA and RNA. That description is correct, but it is too small for modern life science research. In cells, a nucleotide is never just a letter in a polymer. It is also a charged chemical object, a metabolic investment, an enzyme substrate, and, in many systems, a regulatory signal. The same molecular family supports replication, transcription, membrane synthesis, glycan assembly, stress adaptation, and signal transduction. That is why nucleotide biology is not a narrow chapter in biochemistry. It is a control system.
This broader framing matters because the hard questions have changed. Expert readers no longer need a simple reminder that nucleotides contain a base, a sugar, and phosphate. The more useful questions sit deeper. Why is phosphate chemistry so effective at coupling free energy to work? Why do some cells rely heavily on salvage even when de novo synthesis is available? How can a small shift in dNTP ratios destabilize replication fidelity before total nucleotide abundance falls? Why do modified nucleotides behave as both biological marks and analytical targets? And why can one polymerase accept a synthetic nucleotide in one system while another rejects it under nearly identical conditions?
A practical way to organize those questions is to treat nucleotides as operating across three linked layers. The first layer is chemical energetics. This includes charge density, hydrolysis, coupling, and conformational work. The second layer is metabolic flux. Here, cells decide whether to build nucleotides from scratch or recycle them through salvage, while preserving pool balance under changing demand. The third layer is regulatory logic. At this level, nucleotides stop acting like passive metabolites and begin acting like second messengers, stress signals, epigenetic marks, and engineered tools.
That framework helps explain why nucleotide biology often looks simple at first and difficult later. The molecular parts are familiar. The systems behavior is not. A cell can have abundant ATP and still fail to support faithful DNA replication because dNTP ratios are wrong. It can preserve overall purine abundance while losing local repair capacity. It can keep canonical base pairing intact while a methyl modification silently changes recognition by proteins, sequencing platforms, or analytical workflows. The chemistry is modular, but the consequences are system-wide.
To understand those consequences, the chemistry has to come first. This discussion is framed for research-use interpretation of nucleotide chemistry, pathway behavior, and analytical strategy rather than clinical decision-making.
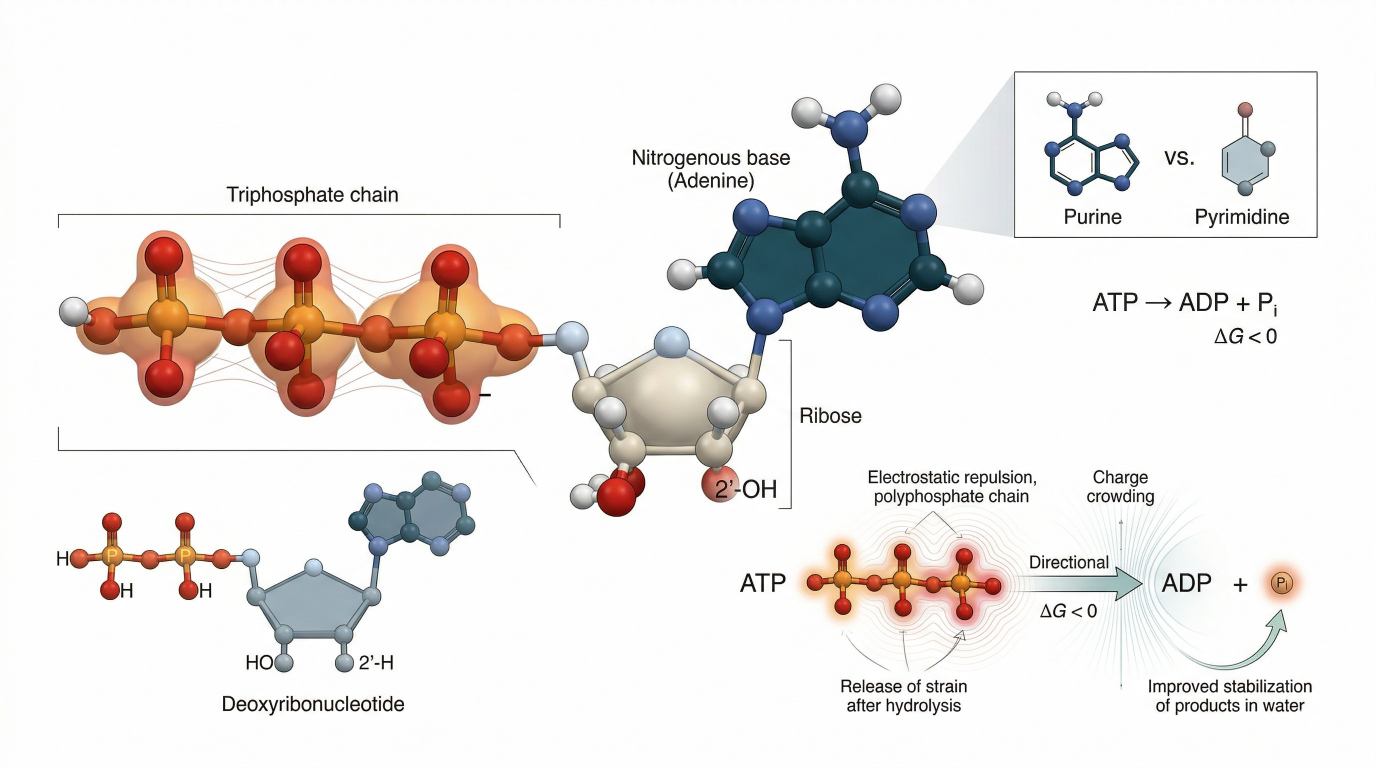 Figure 1: Nucleotide architecture and ATP hydrolysis energetics
Figure 1: Nucleotide architecture and ATP hydrolysis energetics
Modular organization of nucleotides, showing purine versus pyrimidine bases, ribose versus deoxyribose sugars, and the energetic basis of ATP hydrolysis through electrostatic repulsion, resonance stabilization, hydration, and Mg²⁺-dependent context.
Every nucleotide contains three core elements: a nitrogenous base, a pentose sugar, and one or more phosphate groups. That sounds elementary, but the division of labor among those elements explains why nucleotides can do so many jobs without changing molecular family. The base controls pairing logic and recognition surfaces. The sugar alters geometry, flexibility, and chemical reactivity. The phosphate group or phosphate chain controls charge, transfer potential, and energetic coupling. Together they create a modular unit that can be conserved, modified, recycled, or engineered.
The base is the most obvious informational element, but it is also a structural filter. Purines and pyrimidines are not merely two categories of letters. Purines present a larger fused bicyclic scaffold. Pyrimidines use a smaller single-ring scaffold. That size difference affects stacking, steric fit, hydrogen-bond presentation, and how polymerases or repair enzymes interrogate the nucleobase. A purine occupies more space in an active site and presents a different electronic surface. A pyrimidine is more compact and changes the local geometry of recognition. Those differences matter not only for Watson-Crick pairing, but also for how enzymes discriminate between canonical, damaged, and modified bases.
The sugar is equally important, even though it is often treated as background. The difference between ribose and deoxyribose is a single substitution at the 2′ position. Ribose carries a hydroxyl group. Deoxyribose carries hydrogen. That small change reshapes the behavior of the entire polymer. The 2′-OH of ribose increases chemical reactivity and expands structural possibilities. RNA can fold into complex three-dimensional forms, participate in intramolecular reactions, and support transient catalytic or signaling roles because the hydroxyl changes both local geometry and chemistry. DNA lacks that hydroxyl, making it less reactive and better suited for long-term information storage.
That division of labor is not accidental. DNA is chemically quieter because it is built for persistence. RNA is chemically more adventurous because it is built for flexibility. Once phosphate groups are attached, a third layer appears: the ability to store and release free energy in a highly controlled form.
ATP is the standard example, but the phrase "high-energy bond" often creates confusion. Bond breaking does not release energy by itself. Bond cleavage requires input. ATP hydrolysis is favorable because the full reaction produces a more stable product state than the starting state. Several factors contribute. The triphosphate chain contains closely packed negative charge, which creates electrostatic crowding. Hydrolysis relieves part of that crowding. Inorganic phosphate is resonance-stabilized. ADP and phosphate are both well hydrated in water. Charge is distributed more favorably in the products than in the reactant. The result is a negative free-energy change under standard biochemical conditions.
The key idea is not that ATP contains a magical bond. The key idea is that ATP occupies a chemically strained and densely charged state that can be converted into a more favorable one. That is why ATP works so well as a coupling currency. It does not just "contain energy." It links one chemical state to another in a way enzymes can exploit.
Electrostatic repulsion is central to that logic. The phosphate groups of ATP carry substantial negative charge under physiological conditions. Cations such as magnesium help stabilize the molecule, but they do not erase the underlying tension. Hydrolysis reduces that tension and produces species with better stabilization in aqueous solution. The often-cited standard free-energy value for ATP hydrolysis is useful as a reference point, but the real driving force inside cells depends on ATP, ADP, phosphate, magnesium, pH, and local compartment conditions. In practice, ATP is not one universal energetic constant. It is a context-dependent coupling molecule.
That distinction matters because enzymes do not experience ATP as a textbook number. A kinase, ligase, helicase, motor protein, or transporter couples ATP turnover to a very specific sequence of structural events. In one enzyme, the decisive step may be hydrolysis itself. In another, phosphate release may gate the next conformational change. In yet another, ADP release may be rate-limiting. The operational question is therefore not only whether ATP hydrolysis is favorable, but how free energy is partitioned across the reaction cycle.
That same logic extends to the broader nucleotide family. GTP, CTP, and UTP are structurally similar enough to belong to the same energetic universe, yet different enough to be assigned different cellular roles. GTP often functions as a switch-state determinant in signaling proteins. UTP is heavily used in activated sugar metabolism. CTP has a major place in phospholipid biosynthesis. Family resemblance matters, but specialization matters too.
Once that energetic framework is clear, nucleotide metabolism becomes easier to interpret. Cells do not maintain nucleotide pools for one purpose at a time. They must support RNA synthesis, DNA replication, DNA repair, membrane biogenesis, glycan assembly, and signaling at the same time. That means nucleotide abundance is never only a question of supply. It is a question of allocation. A cell can be rich in one pool and fragile in another. It can sustain transcription while approaching a replication fidelity problem. It can preserve gross nucleotide content while failing to support a localized burst of repair or stress signaling.
This is where de novo synthesis and salvage have to be understood as competing flux strategies rather than as two static textbook pathways.
De novo synthesis builds nucleotides from small precursor metabolites. Salvage recovers preformed bases or nucleosides and returns them to the nucleotide economy. At first glance, that sounds like a simple difference between making new material and recycling old material. In reality, it is a deeper difference in metabolic strategy. De novo synthesis is a resource-intensive commitment. Salvage is an efficiency-driven conservation model. The balance between the two reveals how a cell is managing growth, energy, and risk.
Purine de novo synthesis is especially expensive. The purine ring is assembled step by step on an activated ribose platform rather than being synthesized independently and attached later. This requires coordinated inputs from amino acids, one-carbon donors, carbon dioxide, ATP, and multiple enzymes. It is chemically elegant, but metabolically costly. Every step ties nucleotide synthesis to upstream nutrient availability and downstream regulatory control. A cell that leans heavily on de novo purine synthesis is paying for autonomy. It is choosing to generate nucleotide supply even when recycling would be cheaper.
Pyrimidine synthesis follows a different assembly route, but the underlying lesson is the same. Whether the ring is built before attachment to ribose or on the ribose scaffold, de novo synthesis consumes resources that the cell must actively budget. This is why proliferative signaling pathways so often converge on nucleotide biosynthesis. They are not turning on an accessory pathway. They are provisioning the molecular alphabet required for growth.
Salvage looks simpler, but that simplicity is its strength. Instead of reconstructing a base from fundamental precursors, the cell recaptures a usable base or nucleoside and converts it back into a functional nucleotide. That preserves energetic investment, conserves carbon and nitrogen, and can stabilize pools more rapidly than full synthesis. In long-lived or energetically sensitive cell types, salvage is often not just helpful but strategically preferred. Neurons are the classic example. They carry high energetic demand, limited tolerance for waste, and a strong need for stable homeostasis over time. In differentiated cell models, nutrient-limited systems, and maintenance states, salvage logic often becomes even more visible.
That is why salvage should not be treated as a backup route. In many settings, it is the design of choice. Recycling is faster, cheaper, and often less disruptive than rebuilding. This becomes especially clear when one molecule is placed at the center of the discussion: PRPP.
PRPP, or phosphoribosyl pyrophosphate, is often introduced as an activated ribose donor. That is correct, but it is not sufficient. Functionally, PRPP behaves like a network lever. It links pentose phosphate output, growth state, and nucleotide demand. If PRPP availability rises, both salvage and de novo entry become easier. If PRPP falls, competition intensifies. Cells must then choose more carefully which bases are rescued, which biosynthetic routes can remain active, and how much flexibility remains in pool maintenance.
For that reason, PRPP is best understood as a master regulator of nucleotide pool architecture. It is one of the points where broader metabolic state becomes nucleotide state. Carbon flow, proliferative demand, and nucleotide balance meet at this node. A pathway diagram can show PRPP as an intermediate. A systems view shows it as a gatekeeper.
More specifically, PRPP controls both entry efficiency and commitment pressure. On the salvage side, available PRPP determines how effectively free bases can be pulled back into usable nucleotide pools through phosphoribosyltransferase-dependent reactions. On the de novo side, the same activated ribose supply helps determine whether the network can sustain biosynthetic throughput when growth demand rises. This means PRPP is one of the places where pentose phosphate output, precursor competition, and proliferative signaling become experimentally visible as nucleotide behavior. If carbon flow into ribose activation is weak, salvage may become substrate-limited even when bases are present, while de novo routes may become too costly to expand cleanly. If PRPP supply is strong, both routes gain flexibility, but competition between purine and pyrimidine demand still remains. That is why PRPP is not merely a substrate reservoir. It is a control node where metabolic capacity is translated into nucleotide pool architecture.
This flux perspective is far more useful than a static pathway map because real cells do not experience nucleotide metabolism as a clean diagram. They experience it as competing demands across time. A proliferation signal increases the need for nucleotide biosynthesis. Replication stress raises the need for repair-compatible dNTP supply. Nutrient composition changes precursor availability. External nucleosides alter salvage opportunity. Mitochondrial dysfunction can reshape one-carbon support and redox balance. Under each of these conditions, the cell must decide whether to spend more, recycle more, or rebalance differently.
These decisions become most visible at the level of dNTP pool balance.
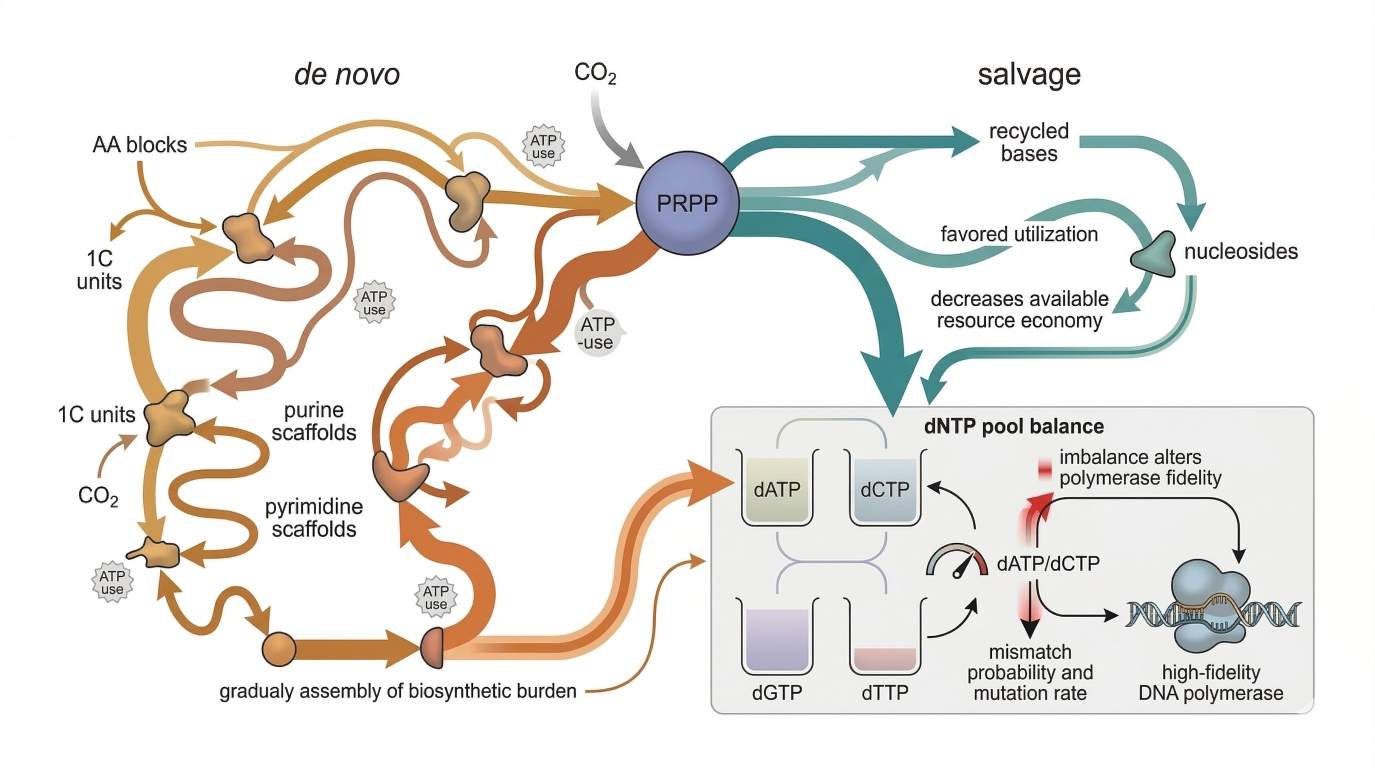 Figure 2: PRPP-centered metabolic flux map of de novo vs. salvage pathways
Figure 2: PRPP-centered metabolic flux map of de novo vs. salvage pathways
PRPP-centered flux architecture showing how carbon input, salvage entry, and de novo synthesis compete to shape nucleotide pool balance, with an inset comparing balanced versus skewed dNTP pools and their impact on replication fidelity.
Researchers often describe dNTP sufficiency as though more substrate is always beneficial until toxicity appears. That is too simple. For DNA replication, total dNTP abundance matters, but relative proportion matters just as much. DNA polymerases do not see a pool as one number. They see four competing substrates, each at a particular concentration, each presented in sequence-specific contexts, and each shaped by local kinetics and proofreading constraints.
A balanced dNTP pool supports rapid and selective incorporation. An imbalanced pool changes the competitive landscape. If one substrate becomes disproportionately abundant, it gains more chances to be sampled, including at positions where it does not belong. What often changes first is not dramatic depletion, but the sampling environment. The polymerase encounters a distorted ratio field before the cell experiences catastrophic shortage. That shift can increase the frequency of near-cognate encounters, enlarge the proofreading burden, and make mismatch discrimination more expensive in kinetic terms. In other words, ratio skew often shows up first as a fidelity pressure problem rather than a simple abundance problem.
The dATP/dCTP ratio is a useful example because it shows the principle clearly. It should not be isolated from dGTP and dTTP, but it captures how ratio control matters. When dATP becomes too high relative to dCTP, the polymerase encounters a different substrate field. Sampling bias increases. Misinsertion pressure can rise in certain sequence contexts. Proofreading demand increases. Extension from marginal pairs may become easier under some conditions because the enzyme is repeatedly challenged by an altered competition landscape. This is why pool imbalance can raise genomic noise even when the total nucleotide pool still appears adequate by bulk measurement.
The exact outcome depends on much more than the ratio alone. Polymerase family matters because active-site geometry and intrinsic selectivity differ. Proofreading capacity matters because exonuclease-proficient enzymes can absorb more error pressure than enzymes with weaker correction. Sequence context matters because local template geometry affects how strongly a mispaired or near-cognate nucleotide is disfavored. Magnesium matters because it reshapes catalytic coordination. Accessory factors and downstream repair matter because some systems correct errors efficiently while others let them persist. For that reason, dNTP imbalance should never be treated as a single-variable predictor of mutation type. It is better understood as a strong upstream bias that alters the probability landscape in which fidelity decisions are made.
This has direct implications for experimental design. In polymerase assays, replication studies, damage-response models, and sequencing library workflows, nucleotide formulation is often treated as a background detail. It is not. It is part of the mechanism. Two systems with the same enzyme and the same template can produce different error profiles if substrate ratios shift. In that sense, fidelity is not merely an intrinsic property of the polymerase. It is an emergent property of polymerase discrimination, substrate competition, proofreading capacity, and repair context under a defined pool condition.
That is exactly why Targeted Metabolomics and Metabolic Flux Analysis (MFA) fit naturally into nucleotide research rather than sitting outside it. Many phenotypes that appear enzyme-driven at first turn out to depend just as strongly on pool composition, pathway routing, or hidden energetic asymmetry once the metabolite layer is measured directly.
Salvage defects illustrate this point well. When salvage efficiency drops, the immediate problem is not always a dramatic fall in total nucleotide abundance. The more subtle problem is asymmetry. Some pools refill faster than others. Some compartments are protected more effectively than others. Some DNA synthesis programs continue under increasingly unfavorable substrate conditions. The cell may still look metabolically viable, yet the fidelity landscape has already shifted. Genome instability can therefore emerge before overt energetic collapse.
The same logic applies when de novo synthesis compensates for poor salvage. Compensation is rarely neutral. Gross nucleotide abundance may recover, but the timing of refill, the cost of maintenance, and the balance between ribonucleotide and deoxyribonucleotide demand can all change. Chemically identical nucleotides can enter the same pool through different routes, yet the route used still changes the surrounding system. Pathway substitution is not always functional equivalence.
This is one reason nucleotide metabolism is so context-sensitive. Growth signaling, nutrient supply, oxygen status, one-carbon metabolism, mitochondrial function, and redox pressure can all shift nucleotide behavior indirectly. A static pathway map cannot capture that. To work at research depth, the investigator has to think in terms of moving constraints rather than isolated reactions.
That systems view also explains why nucleotide pathways are so attractive as experimental intervention points. They sit high enough in cell biology to influence many downstream outputs at once, yet they remain chemically specific enough to measure, perturb, and model. Studies that combine Integrated Proteomics and Metabolomics Analysis with careful pool-level readouts are often better positioned to separate synthesis cost, salvage preference, and stress-induced rewiring than studies relying on endpoint observations alone.
Nucleotides are not only metabolic resources. They are also direct regulatory messengers.
The clearest examples are the cyclic nucleotides cAMP and cGMP. These are not unrelated signaling chemicals. They are structural rewrites of canonical nucleotides into messenger forms. Cyclization changes recognition logic, diffusion behavior, and effector engagement. That conversion turns ATP- and GTP-derived molecules from metabolic currency into fast intracellular instructions.
The signal begins with cyclase activity. Adenylyl cyclases convert ATP into cAMP. Guanylyl cyclases convert GTP into cGMP. The message is then amplified through downstream effectors such as protein kinases, exchange factors, ion channels, and other cyclic nucleotide-binding proteins. But the real sophistication lies in kinetics. Messenger concentration at any moment reflects production, degradation, diffusion, local sequestration, and effector occupancy. The same peak value can mean different things if it appears in a different compartment or persists for a different duration.
This is why cyclic nucleotide signaling cannot be reduced to a simple on-off diagram. Cells generate signaling microdomains, not just global floods of messenger. A local increase in cAMP near one membrane region is not equivalent to a similar increase elsewhere. The molecule is the same. The regulatory meaning is not. Space, timing, and enzyme proximity determine the output.
That is also why Signaling Molecule Analysis can be highly useful in nucleotide-centered projects. A change in second-messenger level becomes much more informative when it is interpreted together with broader metabolic state and downstream response architecture.
The spatiotemporal logic of cAMP and cGMP reveals something deeper about nucleotide biology. The same backbone chemistry that supports metabolism can be reused to encode fast regulatory decisions. A cyclase is not merely an enzyme that makes a messenger. It is a gate that converts one nucleotide state into a signaling regime. A phosphodiesterase is not just a cleanup enzyme. It is a boundary-setting device that determines how far and how long the message can act.
A related but mechanistically distinct logic appears in GTP-binding proteins, where nucleotide state acts as a structural timer. Here, the key difference is not cyclization, but the contrast between GTP-bound and GDP-bound conformations. The terminal phosphate changes how switch regions are stabilized inside the protein. As long as GTP remains bound, one conformation is favored. Once hydrolysis removes the terminal phosphate, the conformational landscape shifts and the signaling state changes.
That is where the next section begins, because GTPases are not just molecular switches. They are timed switches whose chemistry and mechanics are inseparable.
The timing logic of GTPases is what makes them so effective as regulatory devices. ATP is often used to drive directional work. GTP, in many signaling systems, is used to define state. The difference is subtle but important. A GTPase does not simply consume a triphosphate for energy. It uses the presence or absence of the terminal phosphate to stabilize different structural states. The nucleotide is part of the switch architecture itself.
In the GTP-bound state, switch regions align in a way that permits effector engagement. When hydrolysis occurs, the loss of the terminal phosphate changes local contacts and reshapes the active-site environment. The protein relaxes into a different conformational state, usually one with lower signaling output. Guanine nucleotide exchange factors, GTPase-activating proteins, and GDP dissociation regulators then tune how long the switch stays on, how quickly it resets, and how sensitive it is to upstream input. This is why the "GTPase clock" is a useful phrase. The clock is not decorative language. It is a real kinetic feature created by nucleotide-dependent conformational stability.
That clock-like behavior reveals a broader rule about nucleotides. Their role is not limited to being consumed. Often, the nucleotide state itself is the message. The same principle appears again in bacterial stress signaling, where nucleotide derivatives do not just reflect metabolic state. They actively impose it.
One of the clearest examples is ppGpp, the alarmone that drives the stringent response in many bacterial systems. When amino acid supply becomes limiting, ribosome activity is perturbed, or broader nutrient stress appears, cells can shift into a state that favors survival over growth. ppGpp is central to that shift. It rewires transcriptional priorities, dampens biosynthetic expenditure, slows ribosome-associated growth programs, and helps redirect resources away from high-cost anabolism. In other words, ppGpp is not simply a stress marker. It is a nucleotide-derived command signal that reorganizes physiology.
The importance of ppGpp is conceptual as much as mechanistic. It shows that nucleotide chemistry can encode environmental logic. A cell does not only use nucleotides to build macromolecules or fuel reactions. It also uses them to decide whether the current environment justifies continued growth. That is a very different role from ATP hydrolysis or dNTP pool maintenance, yet the underlying language is still nucleotide chemistry. For microbiology, host-microbe systems, and stress-response work, pairing metabolite-level data with Integrated Metagenomic and Metabolomic Analysis can therefore be more informative than measuring transcription alone. Stress state is not only written in genes. It is also written in small-molecule regulators.
Once nucleotides are viewed as regulatory objects, modified nucleotides become much easier to interpret. Epigenetic marks are often described as add-ons to canonical bases. That language is understandable, but it understates the change. A modification such as 5-methylcytosine (5mC) or N6-methyladenosine (m6A) does not replace the base with a new family. Instead, it subtly changes the chemical surface of a familiar base and thereby changes how biology reads it.
That subtlety is exactly why these modifications are powerful. Base pairing can remain largely intact, but recognition by proteins, polymerases, reverse transcriptases, and analytical platforms can shift dramatically. A single methyl group changes hydrogen-bond accessibility, stacking behavior, steric presentation, and sometimes local flexibility. In chromatin biology or RNA regulation, that can be enough to alter recruitment of writers, erasers, and readers. In analytical chemistry, it can be enough to change fragmentation behavior, retention, signal response, ionic current patterns, or enzymatic sensitivity.
This dual identity is what makes nucleotide modification research both elegant and difficult. The molecule is familiar enough to remain part of normal nucleic acid chemistry, but different enough to demand dedicated detection logic. A methylated cytosine is still cytosine-based chemistry. Yet if the research question is site occupancy, absolute abundance, pathway turnover, or coupling to transcriptional outcome, ordinary sequence-level methods are often not enough.
That is where workflow choice becomes decisive. If the main question is global abundance or targeted quantitation of modified nucleosides, mass spectrometry is often the clearest route. Enzymatic digestion reduces nucleic acids to measurable components, and the resulting nucleosides can then be separated and quantified with high sensitivity. For projects focused on bulk modification load, turnover, comparative sample panels, or multiplexed modification profiling, Targeted Metabolomics provides a direct route to chemically grounded measurement.
If the main question is site resolution, the analytical problem changes. Sequencing-adjacent methods become attractive because they preserve positional information. This is where nanopore-based detection has become especially important. Modified bases alter the local signal landscape as a nucleic acid passes through a pore. The platform does not "see" a methyl group directly in the way a structural chemist might, but it does register a changed signal pattern that can be modeled and interpreted. The strength of nanopore detection is not that it eliminates chemistry. The strength is that it allows chemistry to remain attached to sequence context.
That sequence-context advantage matters because modification biology is rarely only about total amount. A small global shift may matter less than a site-specific change at the wrong locus. Conversely, a large total change may be biologically ambiguous if positional information is missing. This is why modern projects often benefit from a layered design in which direct methylation measurement is paired with systems-level interpretation. Bioanalysis of DNA Methylations is useful when the immediate question is modification presence or abundance, while Integrated Analysis of DNA Methylation and Transcriptome becomes more informative when the goal is to connect chemical marking to downstream transcriptional logic.
The same principle applies to RNA modifications such as m6A. The mark itself is chemically small. The biological effects can be large because RNA operates on a short timescale and a dense regulatory surface. Changes in methylation can alter splicing, localization, translation efficiency, or decay. But here again, the challenge is not merely proving that a mark exists. The challenge is deciding what level of measurement actually answers the question. Bulk abundance, transcript selectivity, dynamic turnover, and reader-protein coupling are different problems. They do not collapse into one assay.
This is one place where researchers often underestimate how helpful protein-layer data can be. Modified nucleotides do not act in isolation. Their meaning is created by the proteins that write, erase, bind, or respond to them. For that reason, modification-centered projects often gain a second dimension from Phosphoproteomics Service when signaling-state remodeling is part of the mechanism, or from Integrated Transcriptomic, Proteomic, and Metabolomic Analysis when the goal is to connect nucleotide chemistry to broader regulatory output. The nucleotide mark tells part of the story. The responsive protein network often tells the rest.
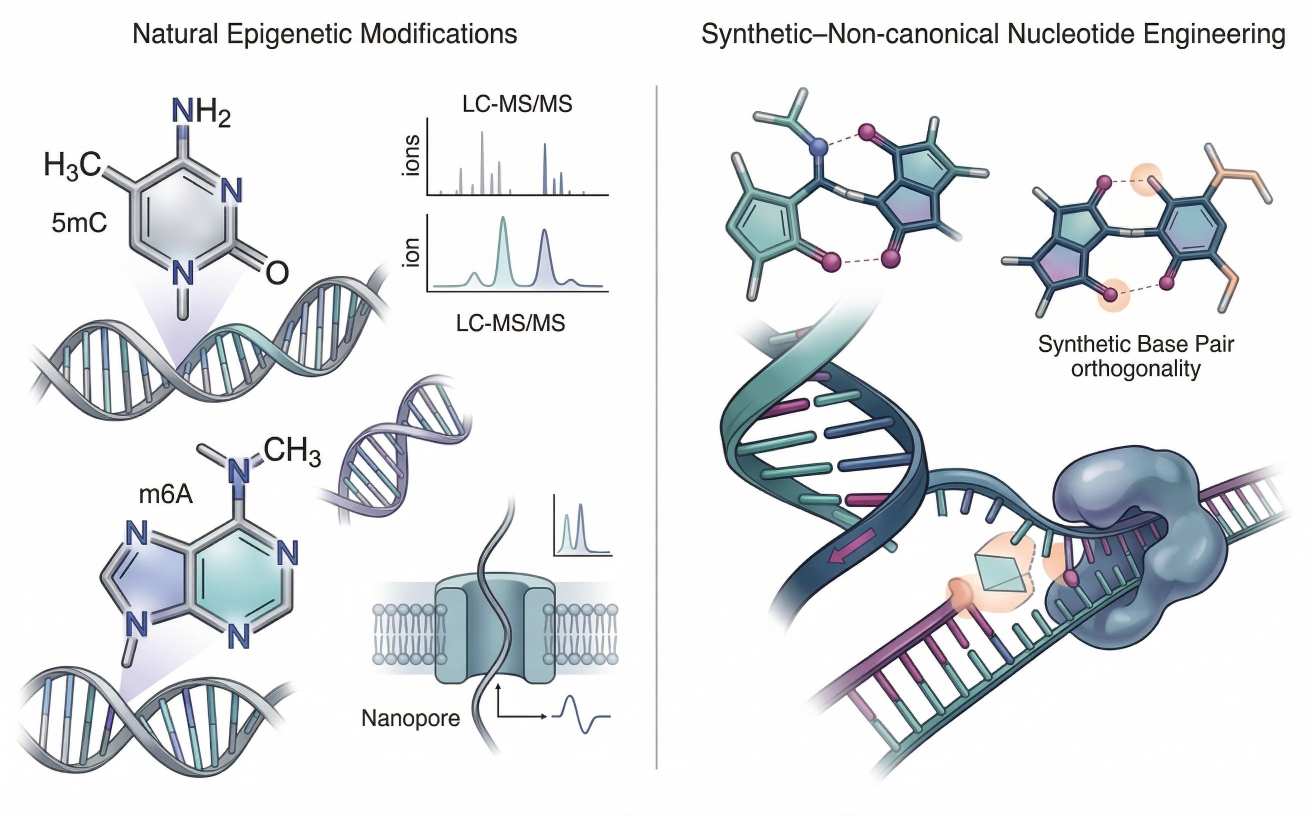 Figure 3: Epigenetic nucleotide modifications and synthetic nucleotide tools
Figure 3: Epigenetic nucleotide modifications and synthetic nucleotide tools
Two-part figure showing natural nucleotide modifications such as 5mC and m6A on one side, and synthetic or non-canonical nucleotide workflows on the other, with analytical panels highlighting mass-spectrometric quantitation, nanopore-based sequence-context detection, and polymerase compatibility constraints.
Once modified nucleotides are framed as chemical signals, the transition to synthetic nucleotides is natural. The question stops being "Can biology tolerate a non-standard base?" and becomes "Under what conditions can a modified or artificial nucleotide preserve enough of the original logic to be useful?" The answer depends on which layer of nucleotide behavior is being challenged.
At the transport layer, the molecule has to reach the right compartment. At the phosphorylation layer, it may need to be converted into mono-, di-, or triphosphate forms efficiently enough to matter. At the polymerase layer, it must either mimic canonical geometry closely or present an alternative geometry that the enzyme can still handle. At the repair layer, it must avoid immediate rejection or excision. At the readout layer, it has to generate interpretable data. Synthetic nucleotide engineering succeeds only when enough of those layers align.
That five-layer framework is useful because each failure mode produces a different experimental signature. If transport fails, the analog may look chemically valid in vitro yet never accumulate where the pathway actually operates. If phosphorylation fails, uptake may appear normal while the active triphosphate pool remains negligible. If polymerase acceptance fails, the analog may be present and activated but rarely incorporated. If proofreading or repair dominates, transient incorporation can occur but retention collapses. If readout fails, the chemistry may work biologically while the dataset still looks ambiguous because the platform cannot distinguish the modified state cleanly. This is why synthetic nucleotide studies often stall not because the concept is weak, but because the wrong failure layer is being measured.
That same logic also explains why non-canonical nucleotides are so useful in research. They are not interesting because they are exotic. They are useful because they reveal where the boundaries of nucleotide logic actually sit. If a synthetic base pair can be recognized, incorporated, copied, and maintained under defined conditions, that means the core informational system is more flexible than a purely canonical view would suggest. If it fails at one stage but not another, the failure still teaches something. It tells us whether the limiting factor is transport, kinase recognition, active-site geometry, proofreading, or structural instability.
A related logic appears in nucleotide analog design for pathway interrogation. Here, the goal is not to extend the alphabet but to probe how selective structural resemblance perturbs pathway handling. The effectiveness of such analogs depends on selective resemblance. They must look close enough to the native substrate to enter the pathway, but different enough to disrupt the next step or expose where discrimination occurs. That is what makes the design intellectually useful. The most informative analogs are not generic blockers. They are selectively processed substrates that reveal where pathway discrimination occurs.
From a research perspective, this has two important implications. First, analog behavior cannot be predicted from structure alone. A molecule that resembles a nucleotide in one diagram may fail because transport is poor, phosphorylation is inefficient, or polymerase discrimination is stronger than expected. Second, analog studies work best when chemistry, metabolism, and protein response are measured together. Projects using DIA Quantitative Proteomics Service alongside pool-level metabolite measurements are often better at separating uptake effects from pathway effects, and pathway effects from downstream cellular adaptation.
The most ambitious version of this field is the expanded genetic code. Here, the goal is not just to perturb biology but to build an informational system with additional coding capacity. Synthetic base pairs and semi-synthetic organisms push directly on the assumption that only the canonical alphabet can support stable heredity and function. The challenge is severe because the system has to do several things at once. A synthetic pair must remain orthogonal enough to avoid confusing canonical pairing logic, yet compatible enough to be carried through replication and expression. Too much similarity creates crosstalk. Too much difference destroys processability.
That tension makes synthetic base pairs a perfect case study in molecular logic. They succeed only when the chemistry respects a narrow balance between novelty and compatibility. The sugar-phosphate backbone still matters. Triphosphate handling still matters. Polymerase fit still matters. The new chemistry does not replace nucleotide logic. It negotiates with it.
There is also an analytical lesson here. Expanded genetic systems are only as credible as the data used to verify them. Was the synthetic nucleotide imported? Was it phosphorylated? Was it incorporated at the intended position? Was it maintained across cycles? Was it transcribed or translated into downstream function? Each question sits at a different layer of evidence. That is why complex synthetic nucleotide projects often benefit from Customized Experiments when a standard assay stack is too generic for the chemistry being tested.
A useful way to close the mechanistic discussion is to compare the major canonical nucleoside triphosphates by role rather than by name. They belong to one family, but cells do not deploy them interchangeably.
| Molecule | Primary energetic or regulatory role | Distinctive systems function | Typical high-value research context |
|---|---|---|---|
| ATP | General coupling currency for phosphorylation, transport, and mechanical work | Dominant energetic intermediate; precursor to cAMP | Bioenergetics, kinase systems, transport ATPases, second-messenger production |
| GTP | State-switch determinant in many signaling proteins | Drives GTPase cycles; precursor to cGMP | Signal transduction, ribosome dynamics, small GTPase regulation |
| UTP | Activated donor logic in carbohydrate metabolism | Supports UDP-sugar formation and polysaccharide or glycan biosynthesis | Glycan assembly, matrix synthesis, cell wall biology |
| CTP | Specialized activation chemistry in biosynthesis | Central to phospholipid synthesis and selected activated intermediates | Membrane biogenesis, lipid remodeling, phospholipid pathway analysis |
The comparison makes one larger point. Nucleotide identity is not only about sequence. It is about role assignment. ATP is favored where flexible energy coupling is needed. GTP is often favored where switch-state logic matters. UTP becomes prominent when activated sugar metabolism is required. CTP appears where membrane-building chemistry demands it. The family is unified by structure, but differentiated by systems use.
That role differentiation also helps explain why experimental strategy has to match the biological question. When the core question is energetic state, ATP abundance alone is rarely enough without context on turnover, phosphate handling, and competing pathway demand. When the core question is flux, isotope tracing and salvage-versus-de novo discrimination are more informative than static abundance. When the core question is regulatory logic, nucleotide measurements should be paired with protein or transcript readouts so that signal generation is not confused with downstream response. In practice, nucleotide systems are easiest to misread when only one layer is measured. They are easiest to understand when chemistry, routing, and response are interpreted together.
That is the larger lesson of nucleotide biology. Cells use the same chemical family to solve three different problems at once. They use nucleotides to store and release free energy. They use them to manage resource allocation through synthesis and salvage. And they use them to encode decisions, from second-messenger cascades to stress adaptation to epigenetic marking. Synthetic biology extends that logic rather than escaping it. Even the most ambitious non-canonical nucleotide systems still have to negotiate with the original rules of charge, geometry, flux, and enzyme recognition.
That is the real reason nucleotides remain so central to life science research. They are not merely building blocks. They are one of the few molecular languages that cells use across chemistry, metabolism, and information at the same time.
How to Study Nucleotide Systems Experimentally
Nucleotide biology is easiest to misread when only one layer is measured. If the main question is energetic state, ATP abundance alone is rarely enough. The more useful readout is the relationship between ATP turnover, phosphate handling, and pathway demand. A stable ATP pool can coexist with severe stress elsewhere in nucleotide metabolism.
If the main question is pathway routing, static abundance is also not sufficient. De novo and salvage pathways can produce similar endpoint pool sizes while operating under very different metabolic costs and control states. In that setting, isotope tracing, pool-ratio interpretation, and flux-centered analysis are more informative than single time-point concentration data.
If the main question is regulatory logic, nucleotide measurement should be paired with downstream response layers. Cyclic nucleotide signaling, nucleotide-dependent conformational switching, and modification-driven gene regulation all become easier to interpret when metabolite data are read together with transcriptomic or proteomic outputs. In practice, nucleotide systems are best understood when chemistry, routing, and response are measured together rather than in isolation.
FAQ
What is the difference between a nucleoside and a nucleotide?
A nucleoside contains a base and a sugar. A nucleotide contains a base, a sugar, and at least one phosphate group. The phosphate is what allows nucleotides to participate directly in energy coupling, signaling, and polymer synthesis.
Why is ATP hydrolysis favorable?
ATP hydrolysis is favorable because the products are more stabilized than the reactant state. Relief of electrostatic repulsion, resonance stabilization of inorganic phosphate, and improved hydration all contribute to the negative free-energy change.
Why do cells use salvage pathways if de novo synthesis exists?
Salvage conserves energy and preserves previously invested carbon and nitrogen. In many cells, especially energy-sensitive or differentiated systems, salvage is a more economical way to maintain nucleotide pools.
Why does dNTP balance matter more than total dNTP abundance?
DNA polymerases do not respond to one pooled number. They operate in a competitive substrate field made up of four distinct dNTPs. That means a cell can appear globally sufficient in nucleotide supply while still creating a distorted insertion environment that raises proofreading burden and mismatch risk. For additional insights into dntps quantification, explore our in-depth resource.
A skewed pool therefore changes fidelity before catastrophic depletion appears. The outcome also depends on polymerase family, proofreading strength, sequence context, repair capacity, magnesium, and accessory factors, which is why ratio imbalance is best treated as a strong upstream bias rather than a single-variable prediction rule.
How are 5mC and m6A typically measured?
Global abundance is often measured by mass spectrometry after nucleic acid digestion. Site-resolved interpretation may involve sequencing-based approaches, including nanopore-linked workflows or other modification-sensitive methods.
What makes a synthetic nucleotide useful rather than merely unusual?
It must work across multiple layers: transport, phosphorylation, enzyme recognition, structural compatibility, and readout. A synthetic nucleotide becomes useful only when enough of the native nucleotide logic is preserved for the experimental system to process it.
Why is GTP often associated with signaling rather than generic energy transfer?
In many proteins, GTP defines a structural state rather than simply providing energy. The presence or absence of the terminal phosphate changes protein conformation and therefore signaling output.
How should researchers study nucleotide biology in complex systems?
The strongest designs usually combine chemical measurement, flux interpretation, and protein- or transcript-level context. Measuring only abundance can miss routing effects, while measuring only downstream response can miss the chemical cause.
A good experimental design starts by deciding which layer is primary: energetics, flux, or regulation. Once that is clear, assays can be chosen to distinguish pool size from pathway choice and pathway choice from downstream signaling or phenotypic consequence.
References
- Buescher JM, Antoniewicz MR, Boros LG, et al. A roadmap for interpreting ^13C metabolite labeling patterns from cells. Current Opinion in Biotechnology. 2015;34:189–201. DOI: 10.1016/j.copbio.2015.02.003
- Lane AN, Fan TWM. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Research. 2015;43(4):2466–2485. DOI: 10.1093/nar/gkv047
- Mathews CK. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nature Reviews Cancer. 2015;15(9):528–539. DOI: 10.1038/nrc3981
- Traut TW. Physiological concentrations of purines and pyrimidines. Molecular and Cellular Biochemistry. 1994;140(1):1–22. DOI: 10.1007/BF00928361
- Breaker RR. Riboswitches and the RNA world. Cold Spring Harbor Perspectives in Biology. 2012;4(2):a003566. DOI: 10.1101/cshperspect.a003566
- Potrykus K, Cashel M. (p)ppGpp: still magical? Annual Review of Microbiology. 2008;62:35–51. DOI: 10.1146/annurev.micro.62.081307.162903
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. DOI: 10.1038/nature11112
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. DOI: 10.1016/j.cell.2012.05.003
- Korlach J, Turner SW. Going beyond five bases in DNA sequencing. Current Opinion in Structural Biology. 2012;22(3):251–261. DOI: 10.1016/j.sbi.2012.04.002
- Malyshev DA, Dhami K, Lavergne T, et al. A semi-synthetic organism with an expanded genetic alphabet. Nature. 2014;509(7500):385–388. DOI: 10.1038/nature13314
- Dien VT, Holcomb M, Feldman AW, et al. Progress toward simple and efficient semi-synthetic organisms. Current Opinion in Chemical Biology. 2018;46:196–202. DOI: 10.1016/j.cbpa.2018.07.010
- Julius C, Yuzenkova Y. Bacterial transcription, translation, and the alarmone ppGpp. Journal of Molecular Biology. 2017;429(20):3285–3301. DOI: 10.1016/j.jmb.2017.08.019









