Introduction
Amino acids are the fundamental building blocks of proteins and peptides, and their accurate identification and quantification form the foundation of protein characterization, nutritional assessment, and biopharmaceutical quality control. Understanding amino acid chemistry is not merely an academic exercise. It is a practical prerequisite for selecting the right analytical method, interpreting data correctly, and troubleshooting common assay failures that can lead to incorrect protein quantification, batch release delays, or regulatory observations. A researcher who understands why tryptophan disappears during acid hydrolysis, why cysteine results are unreliable without performic acid oxidation, or why glutamine and asparagine cannot be distinguished from their acidic counterparts after standard hydrolysis, is far better equipped to design experiments, evaluate analytical results from contract service providers, and avoid costly misinterpretations. The same applies to understanding how side chain chemistry affects detection limits, how to choose the right derivatization strategy, and how to identify common analytical artifacts that can compromise data quality. This guide examines amino acid structure from the perspective of the analytical scientist, connecting molecular properties to detection strategies, messengers, and method development workflows. Each section is designed to provide actionable insights that can be directly applied to the design of analytical methods and the interpretation of amino acid analysis data from contract service providers. The practical focus of this guide distinguishes it from standard textbook treatments of amino acid chemistry, which typically emphasize biological function rather than analytical decision-making and troubleshooting strategies.
Amino Acid Architecture: The Universal Template and the Variable Side Chain
Every standard amino acid shares a common structural backbone consisting of a central alpha-carbon atom bonded to four distinct substituents: an amino group, a carboxyl group, a hydrogen atom, and a variable side chain designated as the R group. The alpha-carbon is a chiral center for all standard amino acids except glycine, where the side chain is simply a second hydrogen atom, making glycine the only achiral proteinogenic amino acid. This chirality has practical significance because most biological systems utilize only L-amino acids, and analytical methods must be capable of distinguishing D and L enantiomers when required for food science, clinical diagnostics, or geochronology applications. The tetrahedral geometry around the alpha-carbon creates two non-superimposable mirror image configurations, designated L and D. Enzymatic systems involved in protein synthesis are stereospecific for L-amino acids, while D-amino acids occur in bacterial cell wall peptides and some bioactive peptides. Distinguishing these forms requires chiral separation methods, typically using chiral stationary phases or chiral derivatization reagents that form diastereomers separable on conventional columns.
Figure 1: Amino Acid General Structure with Functional Group Labels
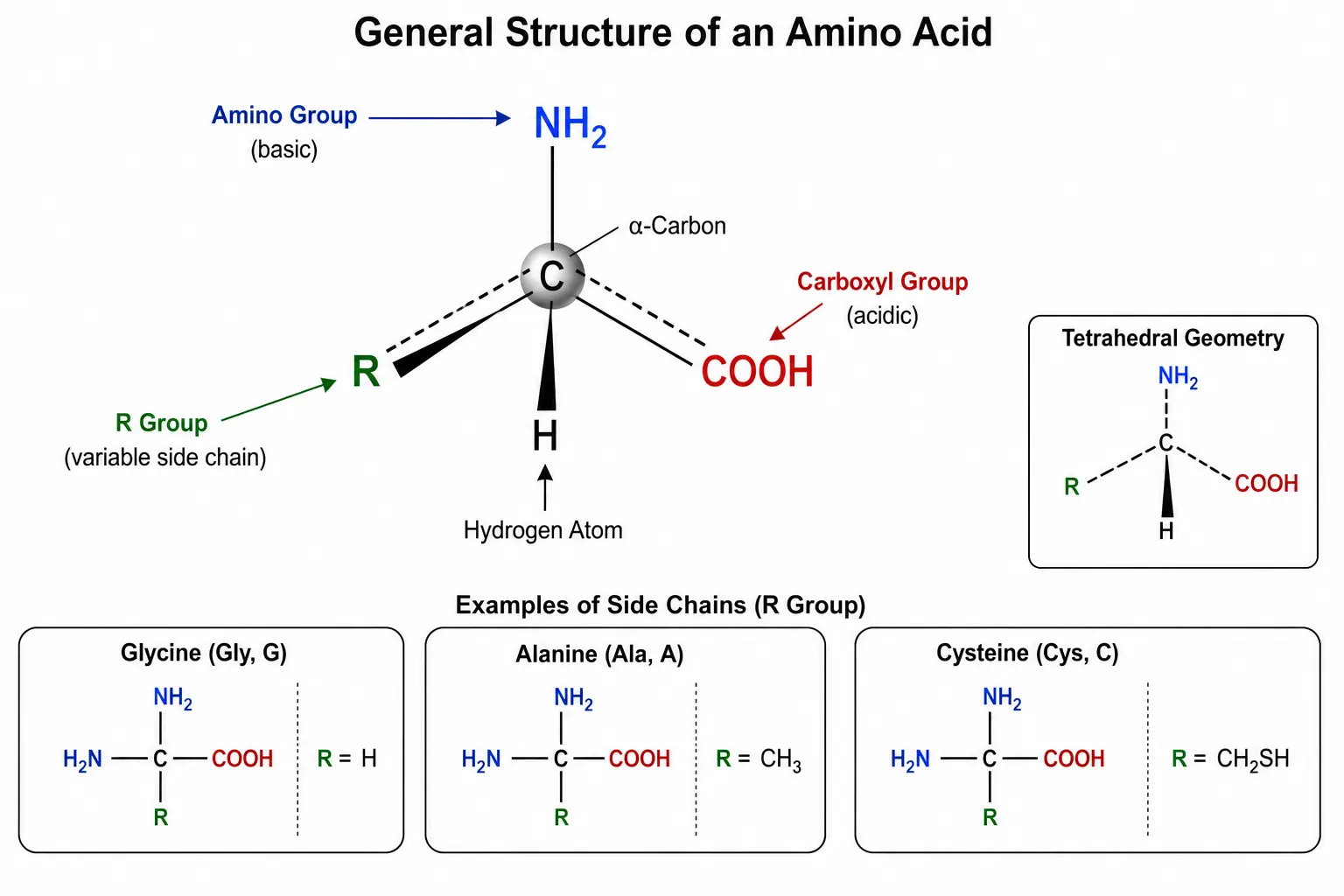
The side chain is the structural feature that differentiates the twenty standard amino acids and determines their chemical behavior. Side chains range in complexity from a single hydrogen atom in glycine to the elaborate indole ring of tryptophan. They include hydrocarbon chains, alcohol groups, thiol groups, carboxylic acids, amides, amines, and heterocyclic rings. Each functional group class imparts specific properties that influence solubility, reactivity, UV absorbance, ionization behavior, and susceptibility to chemical modification during sample preparation and analysis. Understanding these individual properties is essential for method development because each amino acid presents unique analytical challenges. For example, the sulfhydryl group of cysteine is highly reactive and prone to oxidation, the indole ring of tryptophan is acid-labile, and the imidazole ring of histidine has a pKa near neutrality that makes its ionization state sensitive to small pH changes in the mobile phase. Creative Proteomics Amino Acid Analysis (AAA) Service provides comprehensive quantitative and qualitative analysis across all twenty standard amino acids and numerous non-standard variants, using validated methods optimized for each side-chain class.
Side Chain Classification and Its Direct Impact on Detection
Figure 2: Side Chain Classification to Detection Method Mapping Matrix

Nonpolar Hydrophobic Amino Acids
Nonpolar hydrophobic amino acids, including glycine, alanine, valine, leucine, isoleucine, methionine, and proline, lack chromophores that absorb in the ultraviolet range and do not fluoresce natively. They are invisible to UV absorbance detection at wavelengths above 220 nanometers and completely invisible to fluorescence detection without derivatization. For these amino acids, detection typically requires either derivatization with a chromophore or fluorophore or the use of universal detection methods such as charged aerosol detection or evaporative light scattering detection. CAD and ELSD provide near-uniform response across all nonpolar amino acids, making them suitable for quantification without individual response factor calibration. However, ELSD has a limited dynamic range of approximately two orders of magnitude, while CAD provides a dynamic range of three to four orders. Both detectors require volatile mobile phases, which restricts the choice of buffers and ion-pairing reagents. Proline is the only proteinogenic secondary amino acid, and its unique cyclic structure causes it to react differently with many derivatization reagents. OPA does not react with proline at all, while FMOC and AccQ-Tag produce stable derivatives. This distinction is critical when selecting a derivatization method for samples containing significant proline content, such as collagen.
Aromatic Amino Acids
Aromatic amino acids, phenylalanine, tyrosine, and tryptophan, possess conjugated ring systems that absorb ultraviolet light. Tyrosine and tryptophan exhibit strong absorbance at 280 nanometers, which is widely used for protein concentration estimation in solution. Phenylalanine absorbs at 257 nanometers with lower intensity, approximately one-fifth that of tyrosine at the same concentration. Tryptophan also displays native fluorescence with excitation at 280 nanometers and emission at 348 nanometers, providing exceptionally sensitive detection at sub-picomole levels. The fluorescence quantum yield of tryptophan is highly environment-dependent, decreasing significantly in polar solvents and when the residue is exposed on the protein surface. This property is exploited in protein folding studies, where changes in tryptophan fluorescence report on conformational changes. However, tryptophan is completely destroyed during standard acid hydrolysis with six molar hydrochloric acid, so its quantification requires either alkaline hydrolysis with four molar sodium hydroxide at 110 degrees Celsius for 20 hours or direct spectroscopic analysis of intact proteins. This practical limitation means that any method relying on acid hydrolysis will report zero tryptophan regardless of the actual content, a critical fact that must be communicated to researchers submitting samples for amino acid analysis so they can request alternative hydrolysis conditions when tryptophan quantification is needed.
Polar Uncharged Amino Acids
Polar uncharged amino acids, serine, threonine, cysteine, asparagine, glutamine, and tyrosine, contain hydroxyl, thiol, or amide groups in their side chains. These functional groups do not provide useful intrinsic UV absorbance or fluorescence, but they are reactive sites for derivatization chemistry. Cysteine and its oxidized dimer cystine present a particular analytical challenge because cysteine undergoes oxidation during sample preparation and acid hydrolysis, converting to cystine or further to sulfonic acid derivatives. Accurate cysteine quantification requires prior oxidation with performic acid to convert cysteine and cystine to cysteic acid, which is stable and elutes as a single, well-resolved peak in most chromatographic systems.
Acidic Amino Acids
Acidic amino acids, aspartic acid and glutamic acid, carry an additional carboxyl group in their side chains. In reversed-phase chromatography under acidic mobile phase conditions, these amino acids elute very early because their side chains are fully protonated and their polarity is reduced. However, the additional carboxylic acid group also makes these amino acids excellent candidates for anion-exchange chromatography separation, where the negatively charged side chain interacts with positively charged quaternary amine stationary phases. During acid hydrolysis, asparagine and glutamine are completely deamidated to aspartic acid and glutamic acid, respectively. The reported values for aspartic acid and glutamic acid therefore represent the sum of the acid and its amide form. This means that in a typical compositional report, the aspartic acid value includes both aspartic acid and asparagine, while the glutamic acid value includes both glutamic acid and glutamine. Researchers requiring separate quantification of asparagine and glutamine must use enzymatic hydrolysis with asparaginase and glutaminase, or perform the analysis on intact proteins using proteolytic digestion followed by LC-MS/MS. The difference between total glutamate after hydrolysis and pre-hydrolysis free glutamic acid is the glutamine content.
Basic Amino Acids
Basic amino acids, lysine, arginine, and histidine, contain additional basic groups that carry positive charge under acidic conditions. In reversed-phase chromatography with ion-pairing reagents such as trifluoroacetic acid, basic amino acids show strong retention because the ion pairs formed between TFA and the protonated amino groups are highly hydrophobic. However, TFA at concentrations above 0.1 percent can suppress electrospray ionization in LC-MS analysis, reducing sensitivity for these amino acids by 50 to 80 percent compared to mobile phases without TFA. Alternative ion-pairing reagents such as heptafluorobutyric acid provide stronger ion pairing and better retention for basic amino acids but also cause greater MS signal suppression. Formic acid-based mobile phases provide better MS compatibility while maintaining adequate retention, though the chromatographic resolution between basic amino acids may be reduced. For methods requiring both good retention and MS compatibility, pentafluoropropionic acid at 0.05 to 0.1 percent provides a useful compromise. Histidine is unique among the basic amino acids because its imidazole side chain has a pKa of 6.0, meaning its ionization state changes near physiological pH. This property makes histidine sensitive to small pH changes in the mobile phase, and its retention time can shift significantly with mobile phase pH variations of 0.2 pH units or less. Arginine with its guanidino group pKa of 12.48 remains fully protonated across the entire pH range used in reversed-phase chromatography, producing consistent retention behavior but also contributing to strong tailing on silica-based columns.
Acid Hydrolysis: The Bottleneck Every Analyst Faces
Figure 3: Acid Hydrolysis Workflow with Degradation Points Annotated
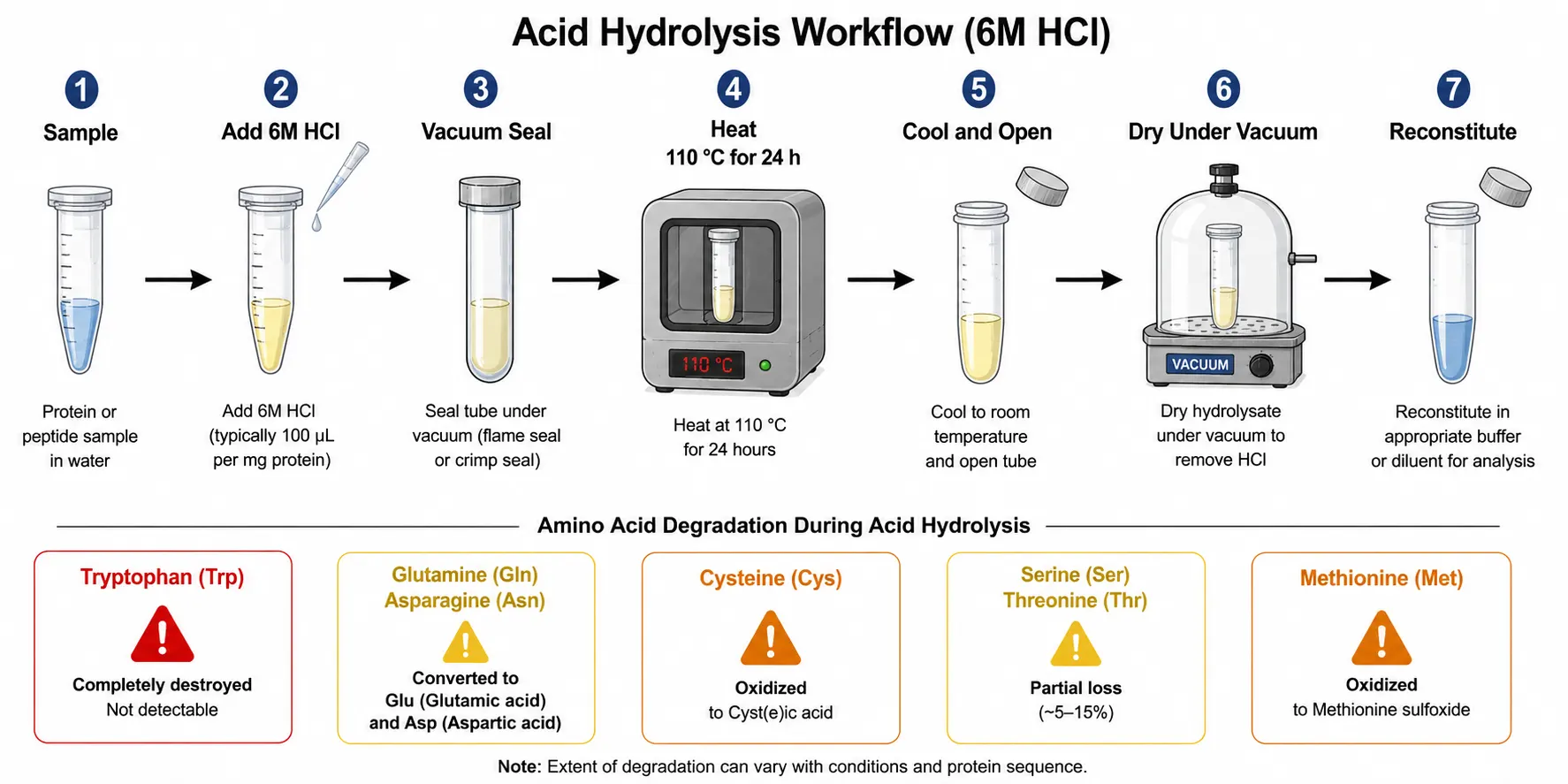
The most common sample preparation method for amino acid analysis is gas-phase acid hydrolysis using six molar hydrochloric acid at 110 degrees Celsius for 24 hours under an inert atmosphere. This protocol was first described in the 1950s and remains the gold standard because it effectively cleaves all peptide bonds while preserving the integrity of most amino acid side chains. Vacuum or nitrogen purging removes dissolved oxygen, which would otherwise promote oxidative degradation during the high-temperature incubation. The sample is typically sealed in a hydrolysis vial or tube under vacuum or inert gas and heated in a block heater or oven. After hydrolysis, the acid is removed by evaporation under vacuum, and the dried hydrolysate is reconstituted in an appropriate buffer for derivatization and analysis. Sample amounts typically range from 5 to 50 micrograms of protein, though LC-MS/MS methods can use as little as 1 microgram. While this protocol is effective for most standard amino acids, every analyst must understand its limitations to avoid misinterpretation of results.
Tryptophan is completely destroyed by acid hydrolysis. The indole ring is cleaved under strongly acidic conditions, and the resulting degradation products do not correspond to any chromatographic peak that would be recognized as tryptophan. The only reliable approach for tryptophan quantification is alkaline hydrolysis using four molar sodium hydroxide at 110 degrees Celsius for 20 hours or direct spectroscopic determination on intact proteins using the absorption at 280 nanometers.
Glutamine and asparagine undergo complete deamidation during acid hydrolysis, converting to glutamic acid and aspartic acid, respectively. The reported glutamate and aspartate values are therefore the sum of the acid and its amide form. Methods reporting separate values for glutamine and asparagine from acid hydrolysis data are simply reporting calculated differences, not direct measurements.
Cysteine and cystine undergo oxidation during hydrolysis. Performic acid oxidation prior to hydrolysis converts both cysteine and cystine quantitatively to cysteic acid, which is stable and produces a single sharp peak in most chromatographic systems. Without performic acid oxidation, cysteine recovery is variable and method-dependent, typically ranging from 30 to 70 percent of the true value.
Serine and threonine undergo partial degradation during extended hydrolysis, losing 5 to 15 percent of their initial content after 24 hours. Valine and isoleucine from hydrophobic protein cores are released slowly, with standard 24-hour hydrolysis achieving only 80 to 90 percent recovery. The most reliable correction for both issues is time-course hydrolysis with sampling at 24, 48, and 72 hours. Degradation follows first-order kinetics, so plotting the natural logarithm of measured serine and threonine against time and extrapolating to zero provides the best estimate of true content. For valine and isoleucine, the measured content increases with time, and a reciprocal plot extrapolated to infinite time provides the correction. This same time-course approach is recommended when the highest accuracy is required for regulatory submissions or reference standard characterization.
Methionine is partially oxidized to methionine sulfoxide during hydrolysis. Quantification of methionine as its sulfoxide derivative requires performic acid oxidation prior to hydrolysis, which converts methionine to methionine sulfone, a stable derivative that is easily quantified.
Derivatization Strategies for HPLC-Based Analysis
Figure 4: Derivatization Reagent Selection Decision Tree
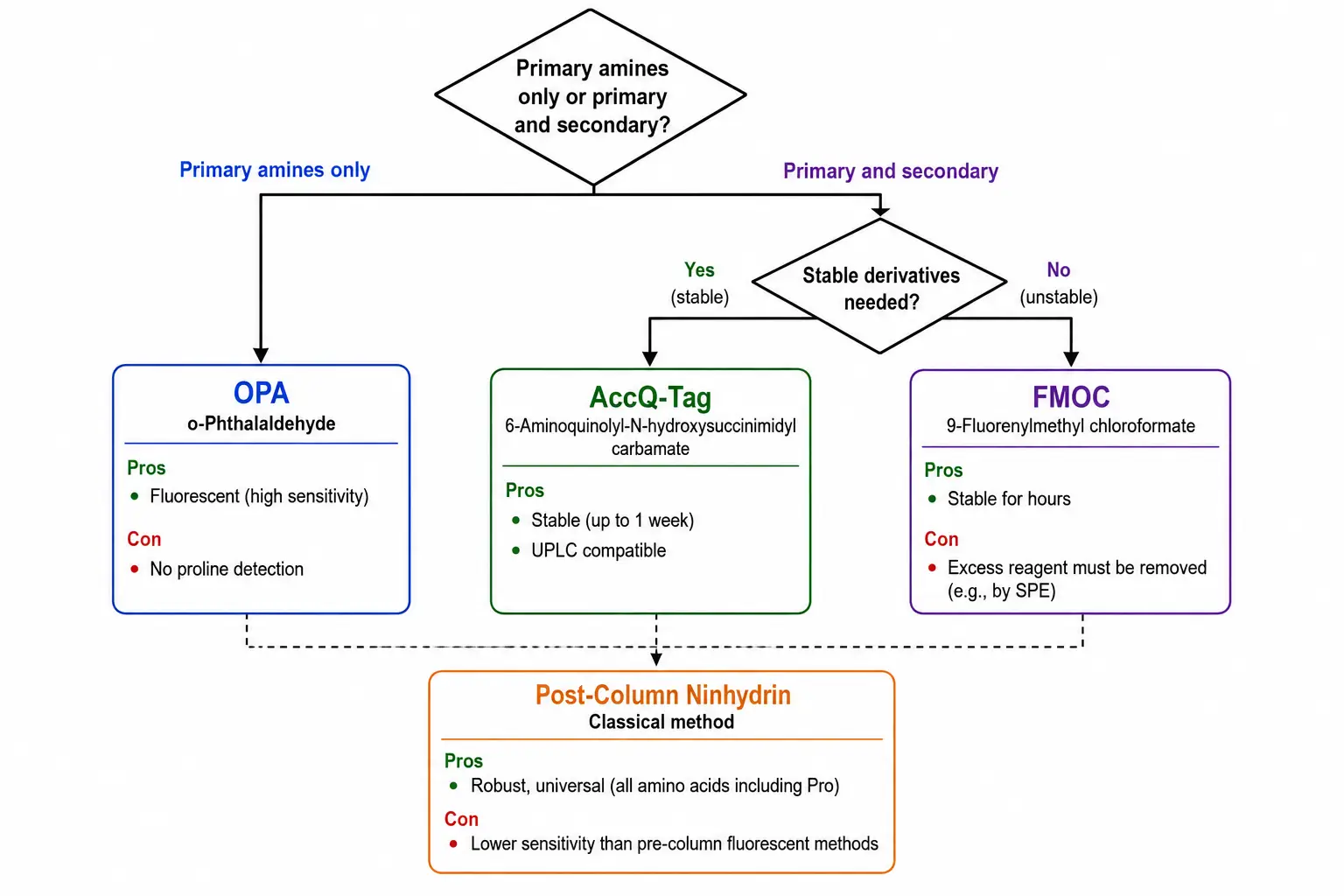
Most amino acids lack intrinsic chromophores or fluorophores at practically useful wavelengths, making chemical derivatization essential for HPLC-based analysis using UV or fluorescence detection. The choice of derivatization reagent determines detection sensitivity, selectivity, chromatographic resolution, and compatibility with downstream detection methods. Each reagent has distinct advantages and limitations that must be matched to the specific analytical requirements. The four most widely used reagents are ortho-phthalaldehyde, fluorenylmethyloxycarbonyl chloride, aminoquinolyl-N-hydroxysuccinimidyl carbamate, and ninhydrin.
Ortho-phthalaldehyde reacts rapidly with primary amines in the presence of a thiol co-reagent such as mercaptopropionic acid to form highly fluorescent isoindole derivatives. OPA derivatives are detected by fluorescence with excitation at 340 nanometers and emission at 450 nanometers, providing excellent sensitivity with detection limits in the low femtomole range. The reaction is complete within minutes at room temperature, making OPA suitable for automated pre-column derivatization. However, OPA does not react with secondary amines such as proline and hydroxyproline, and the isoindole derivatives are chemically unstable, requiring precise timing between derivatization and injection for reproducible quantification.
Fluorenylmethyloxycarbonyl chloride reacts with both primary and secondary amines to form stable fluorescent derivatives. FMOC derivatives are detected by fluorescence with excitation at 260 nanometers and emission at 315 nanometers, with detection limits comparable to OPA. The derivatives are stable for hours, allowing batch derivatization followed by sequential analysis. The excess reagent must be removed by extraction or quenched with a scavenger such as adamantylamine because FMOC reagent peaks can interfere with early-eluting amino acids.
Aminoquinolyl-N-hydroxysuccinimidyl carbamate, marketed as the AccQ-Tag system, reacts with both primary and secondary amines to form highly stable derivatives. AccQ-Tag derivatives are detected by fluorescence with excitation at 250 nanometers and emission at 395 nanometers or by UV absorbance at 260 nanometers. The derivatives are stable for up to one week, making this reagent ideal for unattended batch analysis. The AccQ-Tag Ultra system for UPLC provides complete separation of all 20 standard amino acids plus norvaline and sarcosine internal standards in less than 10 minutes. Pre-column derivatization with AccQ-Tag offers the best compromise between flexibility and reproducibility: the derivatized sample can be injected into any HPLC or UPLC system with fluorescence detection, while the stable derivatives eliminate the timing constraints associated with OPA chemistry.
Ninhydrin is the classical post-column derivatization reagent, reacting with amino acids to produce a purple chromophore detected at 570 nanometers for primary amines and a yellow chromophore at 440 nanometers for secondary amines. While ninhydrin-based systems are robust and well-established, the detection limits are approximately 10 to 50 picomoles, which is 10 to 100 times less sensitive than fluorescence-based methods. The post-column format eliminates the timing issues associated with pre-column derivatization because derivatives are measured immediately after formation. Ninhydrin systems are still widely used in clinical amino acid analyzers for diagnostic applications such as newborn screening for metabolic disorders, where the reproducibility and robustness of the method outweigh the lower sensitivity. The dedicated nature of post-column instrumentation limits flexibility, but the fully automated workflow from injection to report makes it ideal for high-volume clinical laboratories.
Detection Method Selection Based on Amino Acid Properties
Figure 5: Detection Method Sensitivity Comparison Bar Chart
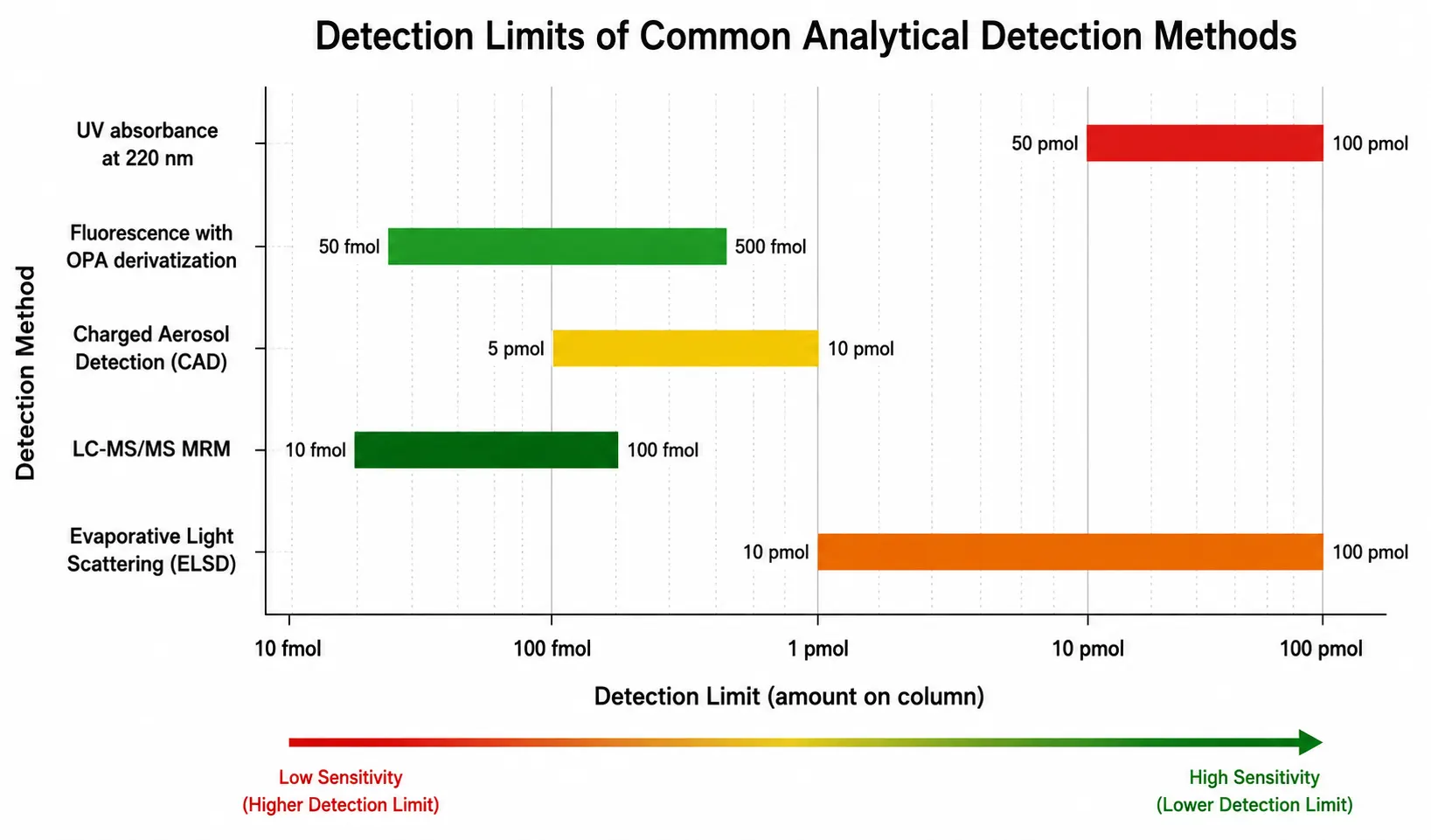
The choice of detection method for amino acid analysis depends on the target amino acids, the required sensitivity, the sample matrix, and the available instrumentation. A practical decision framework can guide method selection: for protein characterization requiring all 20 amino acids with sub-picomole sensitivity, fluorescence detection after AccQ-Tag derivatization provides the best balance of sensitivity, stability, and throughput. For targeted analysis of specific amino acids such as phenylalanine and tyrosine in clinical samples, UV detection may be sufficient and avoids derivatization. For complex biological matrices where matrix effects are a concern, LC-MS/MS with stable isotope internal standards provides the most reliable quantification. For samples where tryptophan and cysteine are critical, parallel alkaline hydrolysis and performic acid oxidation methods are required. UV absorbance detection at 200 to 220 nanometers provides universal detection for all amino acids through the peptide bond chromophore, but sensitivity is poor with detection limits of 50 to 100 picomoles, and many common buffers and mobile phase modifiers absorb strongly in this range. For applications requiring detection limits below one picomole, fluorescence detection after derivatization is the method of choice for most routine applications. Fluorescence methods using OPA or AccQ-Tag achieve detection limits of 50 to 500 femtomoles, which is sufficient for most protein characterization and quality control applications. The main trade-off is the added complexity of the derivatization step, which introduces variability from reaction time, temperature, and reagent quality.
Mass spectrometry detection provides the highest specificity, enabling identification of co-eluting amino acids and detection of modified or non-standard amino acids that would be missed by UV or fluorescence detection. LC-MS and LC-MS/MS methods using hydrophilic interaction chromatography columns separate underivatized amino acids and detect them by selected reaction monitoring, achieving detection limits in the low femtomole range without the variability introduced by derivatization chemistry. HILIC is particularly well-suited for polar metabolites including amino acids because the retention mechanism relies on partitioning between a water-enriched layer on the stationary phase and the organic-rich mobile phase. However, HILIC methods require careful optimization of mobile phase composition, column temperature, and re-equilibration time to achieve reproducible retention times. Matrix effects from co-extracted sample components can reduce ionization efficiency by 50 to 80 percent, and stable isotope-labeled internal standards are essential for accurate quantification in complex biological samples.
Charged aerosol detection and evaporative light scattering detection provide near-universal response for non-volatile analytes, including underivatized amino acids. These detectors are compatible with volatile mobile phases and gradients, making them suitable for method development when sensitivity requirements are moderate, typically 10 to 100 picomoles on-column. CAD offer better sensitivity and wider dynamic range than ELSD, with detection limits approximately 5 to 10 picomoles for most amino acids. Both detectors operate by nebulizing the column effluent into droplets, evaporating the solvent, and measuring the remaining non-volatile particles. The response is independent of the analyte's optical properties, which means that CAD and ELSD can quantify amino acids that lack chromophores without requiring derivatization. The main limitation is that both detectors require strictly volatile mobile phases, excluding non-volatile buffers such as phosphate or borate. Additionally, the response is affected by the organic solvent content of the mobile phase, requiring gradient compensation strategies for accurate quantification in gradient elution methods.
For researchers requiring comprehensive amino acid profiling with the highest sensitivity and specificity, LC-MS/MS with HILIC separation and multiple reaction monitoring provides detection limits below 100 femtomoles for most amino acids, with the additional capability to distinguish structural isomers such as leucine and isoleucine by their fragmentation patterns. Leucine and isoleucine are identical in molecular mass and produce nearly identical derivatives in standard HPLC methods, but their MS/MS fragmentation spectra differ due to the position of the methyl branch, enabling confident individual quantification. For clinical samples where the leucine-to-isoleucine ratio is diagnostically relevant, such as in maple syrup urine disease screening, LC-MS/MS is the only reliable approach. This method also enables detection of modified amino acids, including phosphorylated serine and threonine, acetylated lysine, and methylated arginine, which are not resolved by standard HPLC methods. The trade-off for this enhanced specificity is increased method complexity and longer analysis times for data interpretation. Protein Identification Services integrate amino acid analysis with mass spectrometry-based protein identification and characterization workflows, providing complementary data streams for complete protein characterization.
Practical Applications in Protein Characterization
Figure 6: Sample Preparation to Analysis Workflow Pipeline
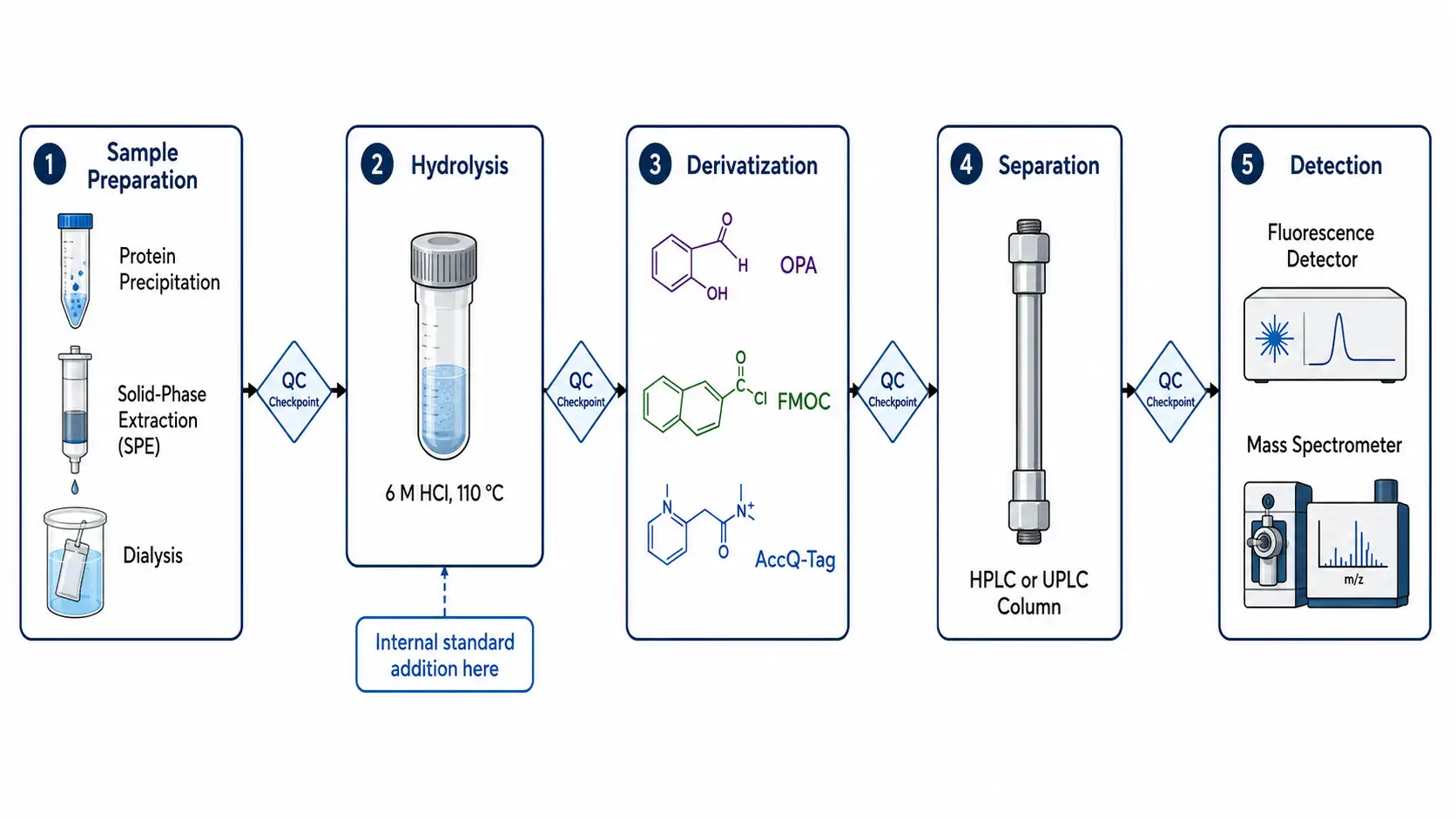
Amino acid analysis serves multiple critical functions in protein characterization throughout the drug development pipeline. The most fundamental application is protein quantification based on amino acid composition. UV absorbance at 280 nanometers estimates protein concentration using the extinction coefficient calculated from the tyrosine and tryptophan content, but this method assumes a known extinction coefficient and native folding. Amino acid analysis provides an orthogonal, absolute quantification method that does not depend on the accuracy of the extinction coefficient or the assumption that all protein is in the native folded state. For biopharmaceutical reference standards, amino acid analysis is the primary reference method for establishing the mass-based concentration of peptide and protein therapeutics. The World Health Organization and regulatory agencies including the FDA and EMA recognize amino acid analysis as a suitable reference method for establishing the peptide content of therapeutic proteins. Under optimized conditions, accuracy of plus or minus 5 to 10 percent is achievable for most stable amino acids, while cysteine, methionine, and tryptophan show higher uncertainty of 15 to 30 percent due to oxidation and degradation during hydrolysis.
For recombinant protein characterization, amino acid analysis confirms the expected composition and detects sequence variants, truncation products, or post-translational modifications that alter composition. A comparison of experimental and theoretical composition provides a score that directly reflects product quality. Compositional agreement within plus or minus 10 percent for all stable amino acids is generally considered acceptable for well-characterized recombinant proteins. For unknown proteins or early discovery samples, the composition can be compared against sequence databases to identify the protein or confirm the presence of expected contaminants. Amino acid composition analysis is also used to calculate the extinction coefficient of a protein, which is essential for accurate UV-based quantification. The theoretical extinction coefficient at 280 nanometers is calculated from the number of tyrosine, tryptophan, and cystine residues, and amino acid analysis provides the experimental confirmation of these values. For monoclonal antibody characterization, amino acid analysis of the reference standard establishes the correct extinction coefficient, which then determines the concentration of all subsequent batches. Discrepancies between theoretical and experimental composition can indicate post-translational modifications, sequence errors, or the presence of non-protein components. These discrepancies are resolved by orthogonal methods such as intact mass analysis or peptide mapping.
Protein purity assessment benefits from amino acid analysis because the technique quantifies total amino acid content, which is proportional to total protein mass. Comparison of total amino acid content with a specific assay such as the bicinchoninic acid or Bradford assay reveals the presence of non-protein components or quantification errors. In biopharmaceutical quality control, amino acid analysis is the reference method for establishing the peptide content standard for therapeutic peptides and proteins.
Post-translational modification analysis leverages the selectivity of amino acid analysis to detect modifications that alter the mass or chromatographic behavior of specific residues. Phosphorylation of serine, threonine, or tyrosine changes their retention time in most chromatographic systems, and phosphorylated derivatives can be detected as additional peaks eluting earlier or later than the unmodified forms. Glycosylation of asparagine or the presence of hydroxyproline in collagen samples can be detected as additional peaks or altered ratios between related amino acids. For monoclonal antibody characterization, amino acid analysis confirms the expected composition of both the heavy and light chains and detects any sequence variants introduced during cell culture or downstream processing. Characterization of Protein Structure combines amino acid composition data with higher-order structural analysis using circular dichroism, differential scanning calorimetry, and hydrogen-deuterium exchange mass spectrometry for comprehensive protein characterization.
Common Pitfalls and Troubleshooting
Figure 7: Common Analytical Artifacts and Solutions Table

Incomplete hydrolysis is the most frequent source of error in amino acid analysis. Hydrophobic peptide bonds involving valine, isoleucine, and leucine are resistant to standard acid hydrolysis conditions and require extended reaction times of 48 to 72 hours for complete release from the protein core. The standard 24-hour hydrolysis typically releases only 80 to 90 percent of these amino acids. Time-course hydrolysis with analysis at multiple time points of 24, 48, and 72 hours, followed by graphical extrapolation to infinite time, provides the most reliable correction for both incomplete release and progressive degradation of acid-labile residues. This approach simultaneously corrects for the loss of serine, threonine, and other acid-sensitive amino acids by extrapolating back to zero time. Interlaboratory reproducibility studies show that experienced laboratories achieve coefficients of variation of 5 to 15 percent for stable amino acids, compared to 15 to 25 percent for labile residues, underscoring the value of validated hydrolysis protocols.
Oxidation artifacts affect methionine, cysteine, and tryptophan. Methionine sulfoxide formation during sample handling can be minimized by working under inert atmosphere and including reducing agents in the hydrolysis buffer. Complete oxidation to methionine sulfone by performic acid treatment provides a stable, quantifiable derivative for total methionine content.
Carryover between injections is a persistent problem in high-sensitivity amino acid analysis. Memory effects from previous injections of high-concentration samples can produce false positive peaks for several subsequent injections, particularly for late-eluting hydrophobic amino acids such as leucine, isoleucine, and phenylalanine. The carryover mechanism involves adsorption of derivatized amino acids to the injection needle, sample loop, or column inlet. Blank injections interspersed every 10 to 20 samples are essential to detect and quantify carryover. When carryover exceeds 0.5 percent of the previous injection, a needle wash protocol using strong organic solvent such as methanol or acetonitrile should be implemented. Thorough column washing protocols with gradient elution to high organic content after each analytical run are essential for maintaining low background and reliable results at sub-picomole detection levels.
Matrix interference from sample components such as salts, detergents, lipids, or carbohydrates can affect hydrolysis efficiency, derivatization yield, and chromatographic resolution. High salt concentrations interfere with hydrolysis by altering the effective acid concentration and can produce unusual chromatographic peaks. Detergents, particularly SDS and Triton, are notoriously difficult to remove and can completely suppress derivatization. Lipids co-extract with hydrophobic amino acids and produce a high background in UV and CAD detection. Sample cleanup by precipitation, solid-phase extraction, or dialysis prior to hydrolysis is essential for complex biological samples such as plasma, tissue homogenates, and cell lysates. The use of isotopically labeled internal standards, added at the earliest possible stage of sample preparation, provides the most reliable correction for matrix effects, recovery losses, and hydrolysis yield variability. Norvaline and sarcosine are the most common non-natural internal standards for HPLC-UV methods, eluting in the middle of the chromatogram without overlapping natural amino acids. For LC-MS methods, deuterium-labeled or carbon-13-labeled amino acids provide superior accuracy because the mass difference ensures complete resolution from the unlabeled analyte. The internal standard must be added before hydrolysis to correct for all subsequent losses.
Frequently Asked Questions
What is the minimum sample amount needed for amino acid analysis?
Five to fifty micrograms of protein is sufficient for standard analysis with fluorescence detection. LC-MS/MS methods can reduce this to one microgram.
How long does a typical amino acid analysis take?
Hydrolysis requires 24 hours, followed by 30 to 60 minutes for analysis. Total turnaround time is typically 2 to 3 business days.
Can amino acid analysis distinguish leucine and isoleucine?
Standard HPLC methods cannot reliably separate these isomers. LC-MS/MS using characteristic fragment ions can distinguish them individually.
Why does my tryptophan result show zero?
Tryptophan is destroyed by standard acid hydrolysis. Alkaline hydrolysis or spectroscopic methods are required.
What is the difference between amino acid analysis and protein sequencing?
Amino acid analysis quantifies total composition. Protein sequencing determines the order of amino acids. They provide complementary information.
How do I choose between pre-column and post-column derivatization?
Pre-column methods offer higher throughput and flexibility. Post-column methods provide better reproducibility using dedicated instrumentation. Stable pre-column reagents such as AccQ-Tag offer the best compromise.
Can amino acid analysis detect D-amino acids?
Yes, using chiral stationary phases or chiral derivatization reagents.
What internal standard should I use?
Norvaline or sarcosine for HPLC-UV methods. Stable isotope-labeled amino acids for LC-MS methods.
How do I correct for hydrolysis losses?
Time-course hydrolysis at 24, 48, and 72 hours with graphical extrapolation.
What is the accuracy of amino acid analysis?
Five to ten percent for stable amino acids. Fifteen to thirty percent for cysteine, methionine, and tryptophan.
References
- Fountoulakis, M., and Lahm, H. W. (1998). Hydrolysis and amino acid composition analysis of proteins. Journal of Chromatography A, 826, 109-134. doi:10.1016/S0021-9673(98)00721-3
- Rutherfurd, S. M., and Gilani, G. S. (2009). Amino acid analysis. Current Protocols in Protein Science, 58, 11.9.1-11.9.37. doi:10.1002/0471140864.ps1109s58
- Xu, W., Zhong, C., Zou, C., Wang, B., and Zhang, N. (2020). Analytical methods for amino acid determination in organisms. Amino Acids, 52, 1071-1088. doi:10.1007/s00726-020-02884-7
- Kambhampati, S., Li, J., Evans, B. S., and Allen, D. K. (2019). Accurate and efficient amino acid analysis for protein quantification using hydrophilic interaction chromatography coupled tandem mass spectrometry. Plant Methods, 15, 46. doi:10.1186/s13007-019-0430-z
- Bjellqvist, B., Hughes, G. J., Pasquali, C., Paquet, N., Ravier, F., Sanchez, J. C., Frutiger, S., and Hochstrasser, D. F. (1993). The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis, 14, 1023-1031. doi:10.1002/elps.11501401163












