Your synthetic peptide arrived. The COA reports 98.2% purity by HPLC. The mass matches within 3 ppm. The sequence is confirmed by MS/MS. You reconstitute, inject into your mouse model, and—every animal in the treatment group develops fever and lethargy. The peptide was pure. It was correctly sequenced. It was also contaminated with endotoxin. Chemical purity says nothing about biological safety. This article explains how to test for what HPLC and mass spectrometry cannot see — and how to ensure your custom synthesized peptide is fit for biologically meaningful experiments.
Why Chemical Purity Is Not Enough
HPLC tells you one thing: what fraction of UV-absorbing material is your target peptide. MS confirms elemental composition and sequence. Neither method detects bacterial endotoxins, live microorganisms, or non-endotoxin pyrogens — contaminants that can turn a well-designed experiment into uninterpretable noise.
Consider a typical scenario. A postdoc treats primary human monocytes with a synthetic peptide at 10 µM. The cells produce a strong TNF-α response. Is the peptide immunogenic, or did endotoxin in the preparation trigger TLR4? Without endotoxin testing data on the COA, there is no way to know. The experiment cannot be interpreted. The peptide — and the weeks spent on the assay — are wasted.
This is not hypothetical. A 2025 investigation of online peptide vendors found 65% of tested products had endotoxin levels exceeding safe thresholds for cell-based work. Even peptides with credible purity and homogeneity data can carry biologically significant endotoxin loads originating from chromatography water, non-depyrogenated glassware, or lyophilization equipment.
The solution is straightforward: request endotoxin and sterility data with every research peptide order. When the vendor cannot provide it, test it yourself. Either way, HPLC purity alone is an incomplete picture of peptide quality.
What Are Endotoxins and Why They Matter
Endotoxins are lipopolysaccharides (LPS) shed from the outer membrane of Gram-negative bacteria. Understanding their structure, stability, and biological activity is the foundation for designing appropriate testing and mitigation strategies.
 Figure 1: LPS Molecular Architecture — Lipid A, Core Oligosaccharide, and O-Antigen Domains Spanning the Gram-Negative Bacterial Outer Membrane
Figure 1: LPS Molecular Architecture — Lipid A, Core Oligosaccharide, and O-Antigen Domains Spanning the Gram-Negative Bacterial Outer Membrane
The lipopolysaccharide monomer consists of three structural domains: a highly conserved Lipid A moiety (the TLR4-active toxic payload) anchored in the outer membrane, a core oligosaccharide bridge, and a variable O-antigen polysaccharide chain extending into the extracellular space.
Endotoxin Structure and Biological Activity
Each LPS molecule consists of three structural domains: a highly conserved lipid A moiety that anchors the molecule to the bacterial membrane, a core oligosaccharide, and a variable O-antigen polysaccharide chain extending outward. The lipid A domain is the toxic payload — it is recognized by Toll-like receptor 4 (TLR4) on mammalian immune cells, triggering a signaling cascade that releases TNF-α, IL-1β, and IL-6.
Endotoxin is remarkably stable. Standard autoclaving at 121°C kills bacteria but does not inactivate LPS. Dry heat at 250°C for 30 minutes or 180°C for 3 hours is required for destruction. A 0.22 µm sterile filter removes intact bacteria but passes endotoxin unimpeded — LPS micelles are far smaller than the filter pores. These properties make endotoxin the most pervasive biological contaminant in peptide research: it survives sterilization, it survives filtration, and it is biologically active at concentrations far below the detection limit of HPLC or MS.
How Endotoxin Contamination Confounds Research
Endotoxin does not merely add noise — it systematically biases experimental outcomes in ways that can masquerade as genuine biological effects.
In cell-based assays, endotoxin at concentrations as low as 0.1 EU/mL induces dendritic cell maturation, upregulates costimulatory molecules, and drives cytokine secretion profiles indistinguishable from a peptide-specific immune response. A 2025 study in Frontiers in Immunology demonstrated that endotoxin levels commonly found in commercial protein preparations (0.1-1.0 EU/mL) significantly altered in vitro T cell proliferation and immunogenicity readouts. The authors recommended that in vitro immunogenicity assays use reagents with endotoxin levels below 0.05 EU/mL — an order of magnitude stricter than many commercial specifications.
In animal models, endotoxin contamination produces fever, lethargy, altered pharmacokinetics, and non-specific immune activation. A single intraperitoneal injection of 0.1 µg LPS (roughly 1,000 EU) is sufficient to induce measurable sickness behavior in mice. For a peptide administered at 10 mg/kg, endotoxin levels of just 0.1 EU/mg translate to biologically active LPS doses in vivo.
The practical consequence: endotoxin testing is not optional for peptides intended for cell-based assays, in vivo studies, or immunological experiments. It is an essential quality parameter.
Endotoxin Testing Methods
Bacterial Endotoxins Testing (BET), as defined by USP<85>and Ph. Eur. 2.6.14, is the compendial framework for detecting and quantifying endotoxin. Three method families dominate: the Limulus amebocyte lysate (LAL) assay in its several formats, the recombinant Factor C (rFC) assay, and the monocyte activation test (MAT). Each has distinct strengths, limitations, and suitability for peptide samples.
LAL — Formats, Sensitivity, and Limitations
The LAL assay uses a clotting enzyme cascade derived from horseshoe crab amebocytes. When endotoxin activates Factor C, a proteolytic cascade produces a gel clot (gel-clot method), a color change (chromogenic method), or turbidity (turbidimetric method). The three formats share the same core biochemistry but differ in readout and quantification.
The gel-clot method is the simplest: serial dilutions of sample are incubated with LAL reagent, and the highest dilution that forms a firm clot determines the endotoxin concentration. It is qualitative or semi-quantitative, inexpensive, and suitable for rapid screening. The chromogenic method uses a synthetic peptide substrate that releases a chromophore upon cleavage by activated clotting enzyme; absorbance at 405 nm provides quantitative results with sensitivity down to 0.005 EU/mL. The turbidimetric method monitors the change in optical density as the clot forms, offering kinetic readout and automation compatibility.
LAL has known limitations relevant to peptide samples. The Factor G pathway in the LAL cascade is activated by (1,3)-β-D-glucan, a common contaminant from cellulose filters and certain chromatography resins, producing false-positive results. Glucan-blocking buffers mitigate this but add complexity. More critically for synthetic peptides, residual TFA from synthesis, certain buffer salts, and extremes of pH can inhibit or enhance the LAL reaction, producing false negatives or inflated results. Every peptide sample must undergo inhibition/enhancement testing — spiking known endotoxin into the sample matrix and verifying recovery within 50-200% — before LAL results can be trusted.
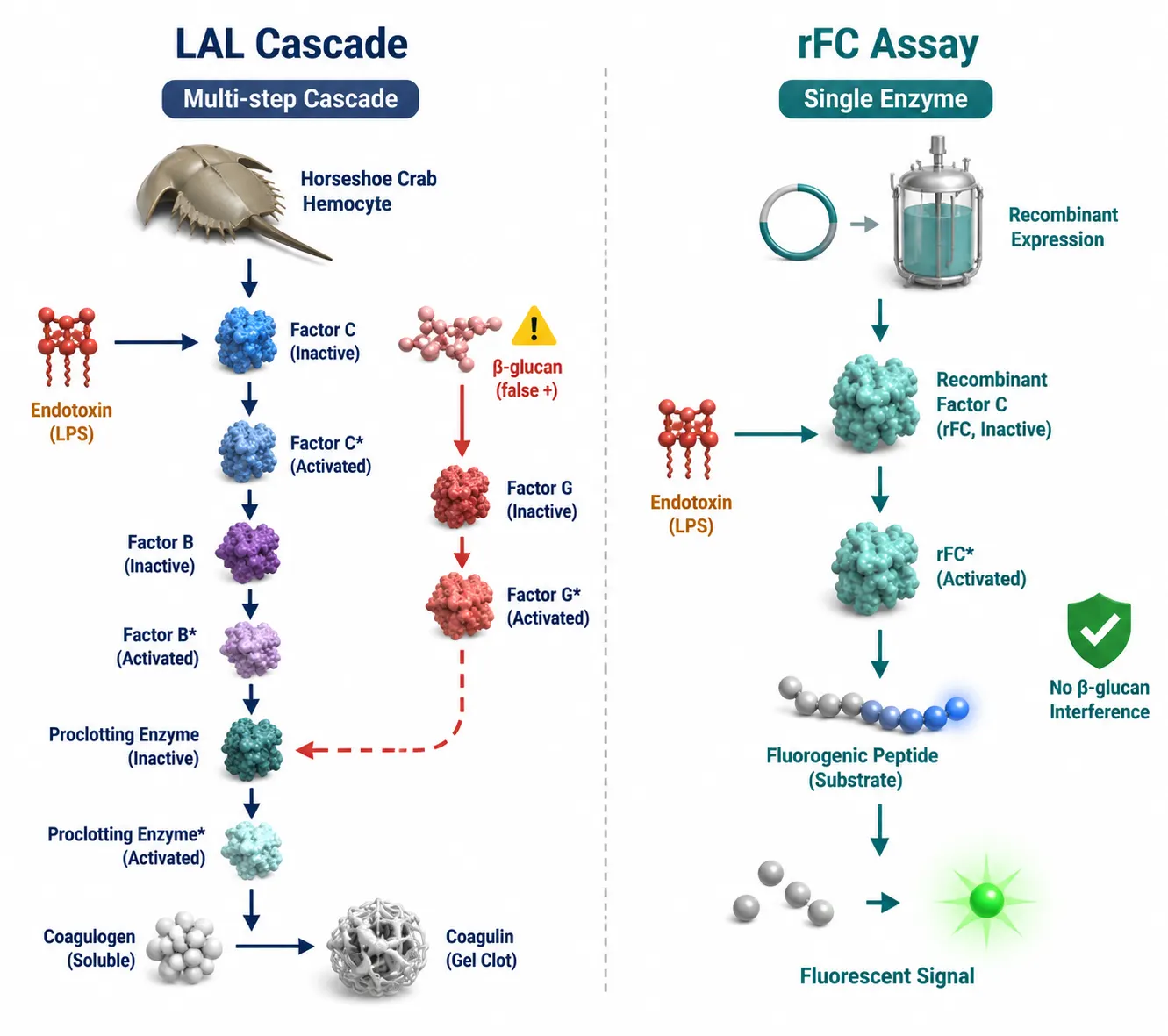 Figure 2: LAL Multi-Step Enzymatic Cascade vs rFC Single-Enzyme Assay — Mechanism Comparison and β-Glucan Interference
Figure 2: LAL Multi-Step Enzymatic Cascade vs rFC Single-Enzyme Assay — Mechanism Comparison and β-Glucan Interference
The LAL cascade (left) involves sequential activation of Factor C, Factor B, and Proclotting Enzyme, with a β-glucan-activated Factor G branch producing false positives. The rFC assay (right) uses a single recombinant Factor C enzyme coupled to a fluorogenic substrate, eliminating the glucan-sensitive pathway.
rFC — The Animal-Free Alternative
The recombinant Factor C (rFC) assay uses a single recombinant protein — Factor C, the endotoxin-activated protease — expressed in a non-animal system, coupled to a fluorogenic substrate. Because rFC bypasses the rest of the LAL cascade, including the glucan-sensitive Factor G branch, it eliminates β-glucan interference entirely.
rFC has achieved significant regulatory milestones. The European Pharmacopoeia will include rFC as Method G in Chapter 2.6.14 by July 2026. USP Chapter<86>already lists rFC as an accepted method, and the FDA has issued guidance supporting rFC use for product release testing. The EDQM published a comprehensive FAQ in January 2026 confirming rFC's equivalence to LAL based on mature, evidence-backed data.
For synthetic peptides specifically, rFC offers advantages beyond regulatory acceptance. bioMérieux demonstrated the ENDOZYME II GO rFC kit with peptide-radiopharmaceutical conjugates at 0.05 EU/mL sensitivity in a 20-minute protocol. ACROBiosystems reported 100% concordance between rFC and multiple LAL kits across 44 sample types including recombinant proteins and injectables. The single-enzyme format reduces the matrix interference surface — fewer enzymatic steps mean fewer points for peptide samples to cause inhibition or enhancement.
The practical recommendation: if endotoxin testing is needed for a research peptide and LAL interference is a concern, rFC is the preferred starting method. It avoids β-glucan false positives, requires less complex interference validation, and is now fully compendial.
MAT — The Full-Spectrum Pyrogen Test
The monocyte activation test (MAT) takes a fundamentally different approach. Rather than measuring enzymatic activation by endotoxin, MAT exposes a human monocytic cell line (typically Mono Mac 6 or cryopreserved PBMCs) to the test sample and measures IL-6 or IL-1β release by ELISA. Because it reads out the actual biological response — the same TLR4, TLR2, and other pattern-recognition receptor pathways activated in vivo — MAT detects the full spectrum of pyrogens: endotoxin (Gram-negative), lipoteichoic acid (Gram-positive), peptidoglycan, fungal pyrogens, and even non-biological pyrogenic particles. This makes MAT the most biologically relevant assay and the only BET method that catches non-endotoxin pyrogens that LAL and rFC both miss. The trade-offs are practical: MAT requires cell culture capability, has higher inter-assay variability (typical CV 20-40% vs 10-15% for chromogenic LAL), and is primarily used when LAL/rFC results are inconclusive or when detection of non-endotoxin pyrogens is critical — for example, peptides destined for in vivo immunological studies where Gram-positive contaminants are also a concern.
Peptide Matrix Interference — Practical Resolution
Synthetic peptide samples present specific challenges for BET. Residual TFA from HPLC purification acidifies the sample and can inhibit the enzymatic cascade. High peptide concentrations can chelate divalent cations required for enzyme activity or physically interfere with substrate access. Organic solvents present in peptide stock solutions (DMSO, acetonitrile) can denature LAL enzymes.
The standard resolution pathway has three steps. First, dilute — most interference is overcome at dilutions of 1:10 to 1:100. Calculate the maximum valid dilution (MVD) from the endotoxin limit and method sensitivity to confirm sufficient headroom for dilution. Second, verify — perform spike recovery at the chosen dilution with a known endotoxin standard; recovery within 50-200% confirms the dilution resolves interference. Third, if dilution is insufficient, switch methods — rFC is less susceptible to many peptide matrix effects than LAL; the monocyte activation test (MAT) provides an orthogonal biological readout that circumvents enzymatic interference entirely.
Endotoxin Limits by Application
Not all experiments carry the same endotoxin risk. The appropriate limit depends on what the peptide will be used for.
For standard cell culture using immortalized cell lines (HEK293, HeLa, CHO), endotoxin levels below 1.0 EU/mg are generally acceptable. These lines are relatively robust; low-level TLR4 activation rarely confounds standard proliferation or transfection readouts.
For primary cell assays — particularly monocytes, macrophages, dendritic cells, or any TLR4-expressing immune cell — the limit should be tightened to below 0.05 EU/mg. Primary immune cells respond to femtogram-level LPS. A 2025 PMC study recommended that in vitro immunogenicity risk assessments use reagents with certified endotoxin below 0.05 EU/mL.
For in vivo rodent studies, limits of 0.1-0.5 EU/mg are typical. The key calculation: if a peptide is dosed at 10 mg/kg to a 25 g mouse, each injection delivers 0.25 mg peptide. At 0.5 EU/mg, the endotoxin dose is 0.125 EU — well below the 0.1 µg (~1,000 EU) threshold for overt sickness behavior. But for immune-focused in vivo studies (vaccine models, tumor immunology), use the tighter 0.05 EU/mg standard.
For therapeutic development — peptides intended for eventual IND-enabling studies or translational research — endotoxin limits follow USP<85>for injectables: 5 EU/kg body weight per hour. For a 70 kg human, this translates to 350 EU per hour. Peptides destined for preclinical regulatory studies should be procured with endotoxin levels below 0.5 EU/mg and tested by a compendial method with full inhibition/enhancement validation.
| Application | Endotoxin Limit | Key Consideration |
|---|---|---|
| Immortalized cell lines (HEK293, HeLa, CHO) | ≤1.0 EU/mg | Low-level TLR4 activation rarely confounds standard assays |
| Primary immune cells (monocytes, DCs, macrophages) | ≤0.05 EU/mg | Sensitive to femtogram-level LPS |
| In vivo rodent studies | 0.1–0.5 EU/mg | Use ≤0.05 EU/mg for immune-focused endpoints |
| Therapeutic development (USP<85>) | 5 EU/kg/hr | Requires full I/E validation with compendial method |
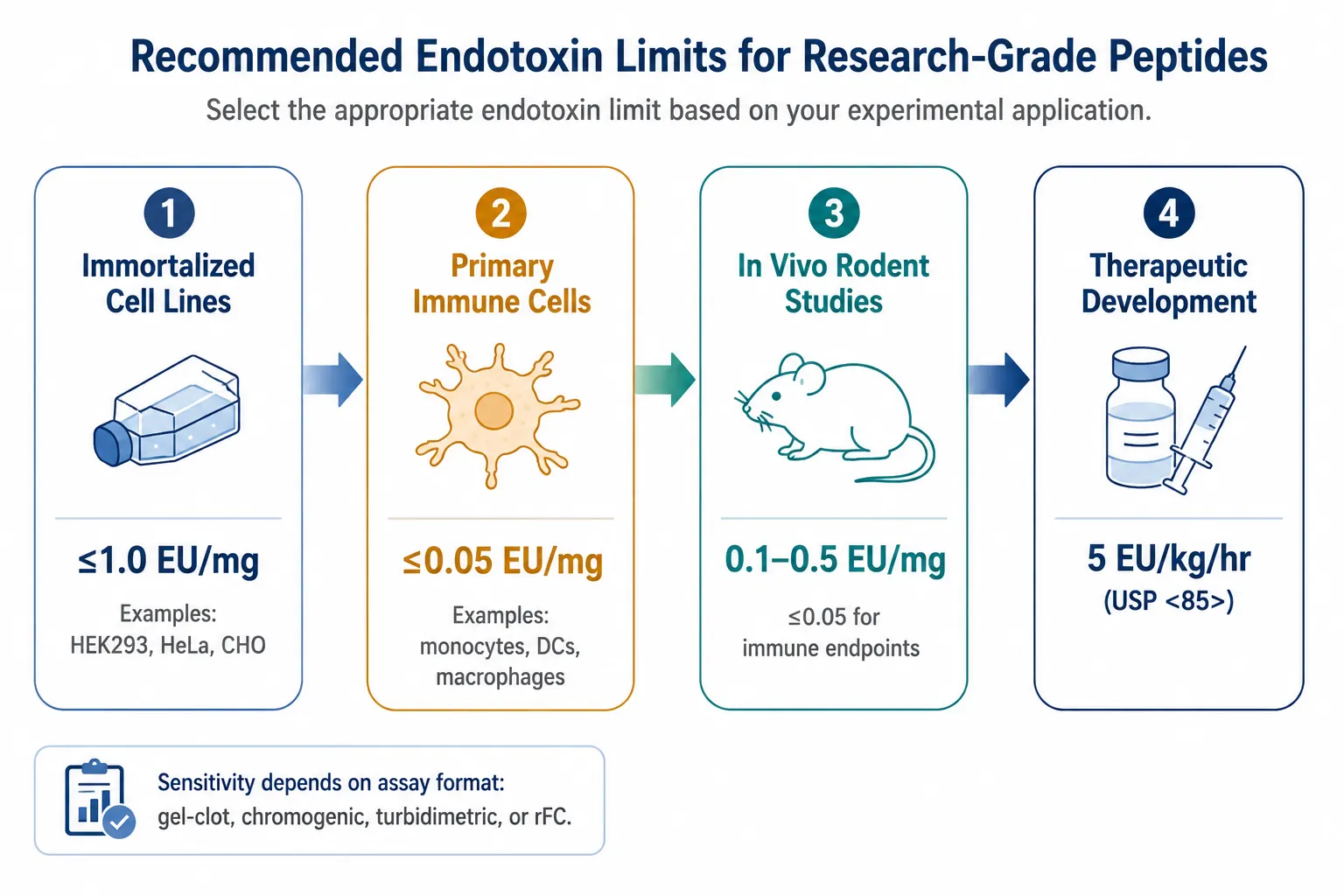 Figure 3: Endotoxin Limit Decision Framework — Recommended Thresholds by Application Type from Standard Cell Culture to Therapeutic Development
Figure 3: Endotoxin Limit Decision Framework — Recommended Thresholds by Application Type from Standard Cell Culture to Therapeutic Development
Endotoxin limits tighten by application: ≤1.0 EU/mg for immortalized cell lines, ≤0.05 EU/mg for primary immune cells, 0.1-0.5 EU/mg for in vivo rodent studies, and 5 EU/kg/hr (USP<85>) for therapeutic development.
Sterility Testing — USP<71>for Research Peptides
Endotoxin testing tells you whether bacterial LPS is present. Sterility testing tells you whether live microorganisms are present. They answer different questions, and both matter.
Membrane Filtration vs Direct Inoculation
USP<71>specifies two methods. Membrane filtration is the gold standard for aqueous peptide solutions. The sample passes through a 0.45 µm filter that traps microorganisms; the filter is rinsed to remove antimicrobial residues, then transferred into two culture media: Fluid Thioglycollate Medium (FTM) incubated at 30-35°C for anaerobic and facultative organisms, and Soybean-Casein Digest Medium (SCDM) at 20-25°C for aerobic bacteria and fungi. Both are incubated for a minimum of 14 days with periodic inspection for turbidity.
Membrane filtration is preferred for peptides because it concentrates the entire sample volume onto a single membrane, maximizing sensitivity. The rinse step removes residual TFA, acetonitrile, or peptide itself — any of which could inhibit microbial growth and produce a false-negative result.
Direct inoculation — adding the peptide solution directly to culture media — is reserved for non-filterable formulations or very small sample volumes. It carries higher false-negative risk because the peptide or its formulation components may suppress microbial growth. Bacteriostasis/fungistasis (B/F) validation — demonstrating that six compendial challenge organisms can grow in the presence of the product — is mandatory before direct inoculation results can be accepted.
For research-grade peptides, membrane filtration with B/F validation is the appropriate standard. If the vendor reports a sterility test result, ask which method was used and whether B/F validation data is available.
Bioburden vs Sterility — A Critical Distinction
Bioburden testing quantifies the total aerobic microbial count and total yeast/mold count before terminal sterilization. Sterility testing confirms the absence of viable microorganisms after sterilization. They are sequential, not interchangeable. A peptide with a bioburden result of "<10 CFU/g" is not sterile. It has simply had its microbial load measured. Sterility requires a 14-day incubation with no growth in either medium.
For research-grade peptides intended for cell culture or in vivo use, sterility claims on the COA should be verified. Not all vendors perform USP<71>sterility testing on "research-grade" products — many use terms like "sterile-filtered" which means only that the solution passed through a 0.22 µm filter, not that a compendial sterility test was performed afterward. For peptides requiring residual contaminant testing alongside sterility, confirm the full panel of quality assays with the vendor.
Sterility Does NOT Equal Endotoxin-Free
This principle is one of the most under-communicated in peptide characterization. A sterile peptide — one with no detectable viable microorganisms — can carry substantial endotoxin. Three mechanisms explain why.
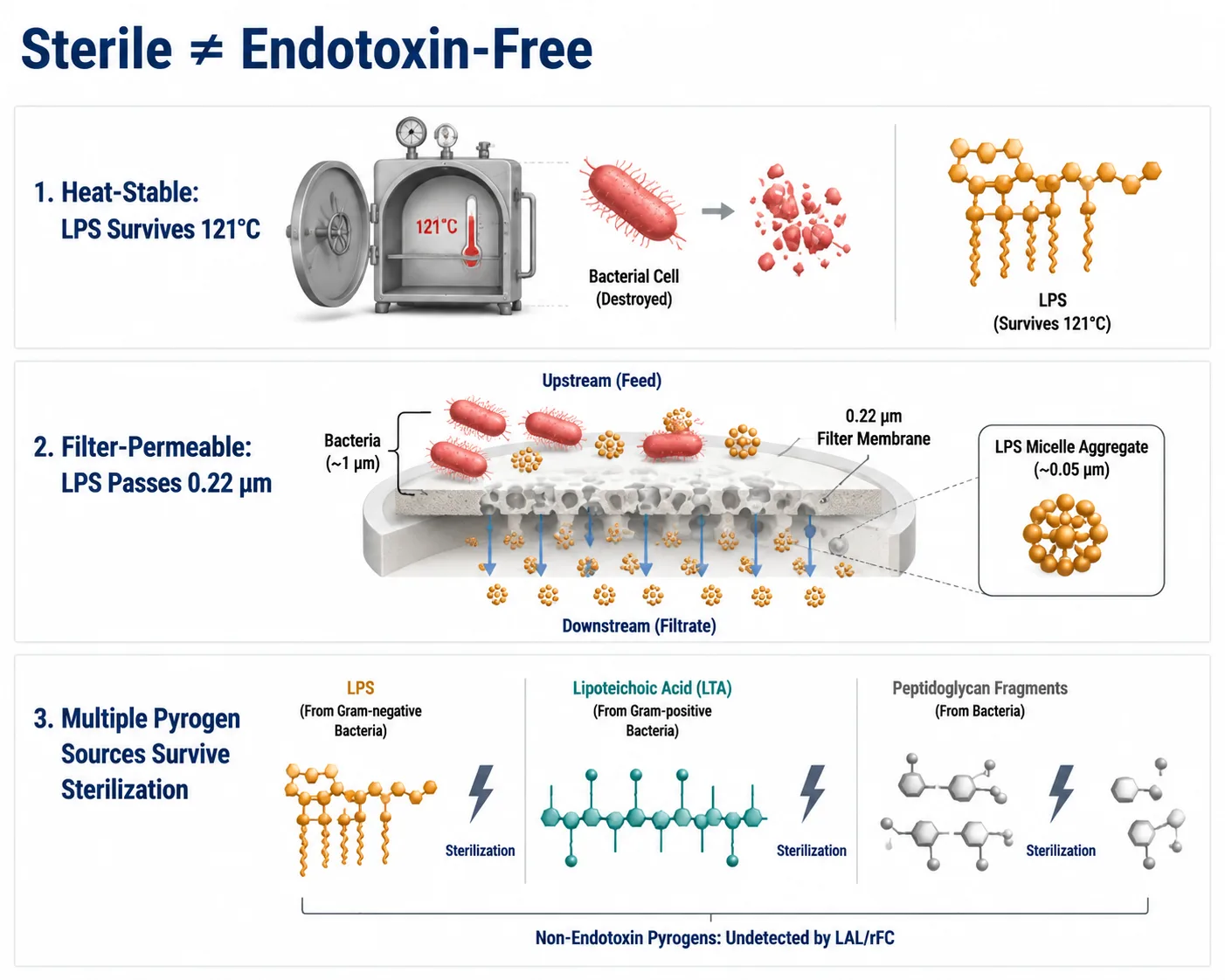 Figure 4: Sterility Does Not Equal Endotoxin-Free — Three Mechanisms Explaining Why Sterile-Filtered Peptides Can Retain Pyrogenic Activity
Figure 4: Sterility Does Not Equal Endotoxin-Free — Three Mechanisms Explaining Why Sterile-Filtered Peptides Can Retain Pyrogenic Activity
Endotoxin survives standard autoclaving (121°C), passes through 0.22 µm sterilizing-grade filters, and non-endotoxin pyrogens (LTA, peptidoglycan) survive sterilization but are undetected by LAL and rFC assays.
First, endotoxin is heat-stable. Autoclaving at 121°C for 15 minutes — the standard sterilization cycle — kills bacteria but leaves LPS intact and biologically active. The bacteria die; the pyrogen remains.
Second, endotoxin passes through sterilizing-grade filters. A 0.22 µm membrane retains intact microorganisms. It does not retain soluble LPS micelles, which range from 300 to 1,000 kDa in aqueous solution — far smaller than the filter cutoff. Sterile filtration removes bacteria. It does not remove endotoxin.
Third, non-endotoxin pyrogens from Gram-positive bacteria — lipoteichoic acid (LTA), peptidoglycan — also survive standard sterilization. These pyrogens are not detected by the LAL or rFC assays. Only the monocyte activation test (MAT) or the rabbit pyrogen test captures the full spectrum of pyrogenic contaminants.
The practical implication: a COA reporting sterility but lacking endotoxin data provides only partial assurance of biological safety. For peptides intended for cell-based or in vivo work, both parameters must be confirmed independently.
Contamination Sources and Prevention
Endotoxin and microbial contamination can enter at any point from synthesis to injection. Understanding the entry points is the first step toward prevention.
Manufacturing-Side Sources
During peptide synthesis, endotoxin can be introduced through non-sterile water used in HPLC mobile phases — water is the single largest-volume raw material in peptide purification, and even WFI-grade water can accumulate endotoxin if distribution lines develop biofilms. Chromatography resins and columns that are not depyrogenated between runs carry residual LPS from previous purification cycles. Lyophilization chambers that are not regularly sanitized can harbor bacterial residues that shed endotoxin onto the dried peptide cake. Glassware and tubing cleaned but not depyrogenated — autoclaving alone is insufficient — contribute cumulative contamination across the process.
A vendor with a documented depyrogenation program, endotoxin-tested water systems, and batch-specific BET data on their COA provides measurably lower-risk material than one offering only HPLC purity and mass confirmation. This is particularly relevant for host cell protein and process-related impurity control, where endotoxin is one of several manufacturing-derived contaminants requiring monitoring.
Laboratory-Side Sources During Reconstitution and Handling
Contamination does not end at manufacture. The most common laboratory introduction routes are:
Non-sterile reconstitution water is the leading cause. Even ultrapure water from a lab system can accumulate endotoxin in distribution lines. Use commercially supplied sterile, endotoxin-tested water for injection (WFI) or certified endotoxin-free water, not water from the lab tap or Milli-Q system.
Aerosol generation during pipetting spreads contaminants. Aggressive pipetting generates microscopic droplets carrying bacteria or endotoxin between vials. Use filtered pipette tips and pipette slowly.
Repeated freeze-thaw cycles compromise the container seal. Each thaw introduces environmental exposure. Aliquot peptides immediately after initial reconstitution into single-use volumes.
Reusable glassware and non-depyrogenated plasticware harbor LPS. LPS adsorbs strongly to glass and plastic surfaces. Items labeled "sterile" are not necessarily endotoxin-free. For sensitive immune assays, use certified pyrogen-free consumables.
A 2025 report documented that recirculated, sterilized glass pipettes previously used for bacterial work induced IL-6 upregulation over 75,000-fold in immune cells — equivalent to adding 1 µg/mL of purified LPS — demonstrating that routine washing and autoclaving are insufficient to remove pyrogenic residues.
Depyrogenation Strategies
Depyrogenation is the destruction or removal of endotoxin. The method choice depends on the target — equipment surfaces, water, or the peptide solution itself.
 Figure 5: Depyrogenation Strategy Selection Flow — Four Pathways Categorized by Target Material from Equipment to Peptide Solutions
Figure 5: Depyrogenation Strategy Selection Flow — Four Pathways Categorized by Target Material from Equipment to Peptide Solutions
Depyrogenation methods are target-specific: dry heat (250°C/30 min) for equipment and glassware, ultrafiltration (10 kDa MWCO) for water and aqueous buffers, strong anion exchange chromatography (flow-through mode) for peptide solutions, and Triton X-114 phase separation for small-scale research applications.
For equipment and glassware, dry heat is definitive. 250°C for 30 minutes or 180°C for 3 hours inactivates endotoxin by oxidizing the lipid A moiety. This is the method specified in USP<85>and Ph. Eur. 5.1.7 for depyrogenation of containers and utensils. 1 N sodium hydroxide treatment with extended contact time (>1 hour) is an effective room-temperature alternative for chromatography columns and tubing that cannot withstand dry heat.
For water and aqueous buffers, ultrafiltration with a 10 kDa nominal molecular weight cutoff membrane retains endotoxin aggregates (300-1,000 kDa) while allowing water, salts, and small-molecule buffers to pass. This is how WFI systems control endotoxin. It is not suitable for peptide solutions, because the peptide itself would be retained or partially retained.
For peptide solutions, ion exchange chromatography is the preferred method. At neutral pH, endotoxin carries a net negative charge from phosphoryl and carboxyl groups on the lipid A and core oligosaccharide. A strong anion exchange membrane (quaternary ammonium, Q-type) in flow-through mode — with the buffer pH adjusted below the peptide's pI — binds endotoxin while allowing the positively charged peptide to pass unretained. Sartorius Sartobind Q membranes have demonstrated over 3 log reduction values with greater than 95% protein recovery under optimized pH conditions. After depyrogenation, Amino Acid Analysis (AAA) can confirm the peptide's composition was not altered by the treatment.
For small research quantities where chromatography is impractical, Triton X-114 phase separation offers a low-throughput alternative. The peptide solution is mixed with Triton X-114 at 4°C, warmed to 37°C to induce phase separation, and centrifuged. Endotoxin partitions into the detergent-rich phase; the aqueous phase containing the peptide is recovered. Multiple cycles improve clearance. This method is effective but requires subsequent detergent removal and is not suitable for all peptides.
The choice of depyrogenation strategy depends on what you are treating and at what scale. Dry heat for equipment. Ultrafiltration for water. Anion exchange for peptide solutions. Triton X-114 for small-scale research applications where chromatography equipment is unavailable.
FAQ
Q: Do I really need both sterility AND endotoxin testing for my peptide?
Yes. Sterility testing confirms the absence of live bacteria. Endotoxin testing confirms the absence of the pyrogenic LPS molecules that bacteria leave behind. A sterile peptide can carry biologically significant endotoxin. Both parameters matter for cell-based and in vivo work.
Q: What endotoxin level is acceptable for cell culture?
For standard immortalized cell lines, below 1.0 EU/mg is generally sufficient. For primary immune cells (monocytes, macrophages, dendritic cells), aim for below 0.05 EU/mg. For in vivo studies, 0.1-0.5 EU/mg is typical unless the study focuses on immune endpoints, in which case use the stricter limit.
Q: Should I use LAL or rFC for my peptide?
Start with rFC if it is available. It avoids β-glucan false positives, requires simpler interference validation, and is now compendial in both USP<86>and Ph. Eur. 2.6.14. Use LAL if your peptide contains components that specifically interfere with rFC or if regulatory precedent requires it.
Q: Can I perform sterility testing in my own lab?
Technically yes, but the practical barriers are significant. USP<71>sterility testing requires a Grade A biosafety cabinet or isolator, validated media, compendial challenge organisms for method suitability, and a 14-day incubation period. For most research labs, requesting sterility testing from the peptide vendor or a contract testing laboratory is more practical.
Q: What causes false negatives in sterility testing of peptides?
Residual antimicrobial activity from the peptide itself or from formulation components (TFA, organic solvents, low pH) that suppress microbial growth. This is why bacteriostasis/fungistasis validation with six compendial challenge organisms is mandatory before sterility test results can be accepted for a given peptide formulation.
Q: Does sterile filtration remove endotoxin?
No. A 0.22 µm filter removes intact bacteria but does not retain soluble LPS aggregates, which range from 300 to over 1,000 kDa and pass through sterilizing-grade filters unimpeded. Endotoxin must be removed by dry heat (equipment), ultrafiltration (water), ion exchange chromatography (peptide solutions), or phase separation — not by filtration.
Q: What is the single most cost-effective step to reduce endotoxin risk in my peptide experiments?
Use commercially supplied sterile, endotoxin-tested water for reconstitution instead of lab water, and reconstitute in a biosafety cabinet using certified pyrogen-free consumables. A substantial fraction of laboratory endotoxin problems originate at the reconstitution step, and this change costs less than repeating one failed experiment.
References:
- Berdnikova ZE, Tikhonova AS. Recommendations for the development of sterility testing procedures for biological medicinal products based on pharmacopoeial methods. Biological Products. Prevention, Diagnosis, Treatment. 2024;24(2):229–236. doi:10.30895/2221-996X-2024-24-2-229-236.
- Baker E, et al. Barriers to the use of recombinant bacterial endotoxins test methods in parenteral drug, vaccine and device safety testing. Alternatives to Laboratory Animals. 2023;51(6):401–410. doi:10.1177/02611929231204782.
- Kelley M, Stevens I, Marchessault N, Akiyoshi J, Jahngen EG. Evaluation of PyroSmart NextGen® recombinant cascade reagent for bacterial endotoxin testing. BPB Reports. 2023;6:11–15. doi:10.1248/bpbreports.6.1_11.
- Klein K, Heisterberg J, Stolk P. Synthetic polypeptides using a biologic as a reference medicinal product — the European landscape of regulatory approvals. Frontiers in Medicine. 2024;11:1335928. doi:10.3389/fmed.2024.1335928.
- Jeong YH, Lennon G, Veldman G, Serna DM, Ibrahimov A. Establishing endotoxin limits to enhance the reliability of in vitro immunogenicity risk assessments. mAbs. 2025;17(1):2458627. doi:10.3390/ijms26010015.
- Park H, et al. A Study on the Application of Recombinant Factor C (rFC) Assay Using Biopharmaceuticals. Microorganisms. 2024;12(3):516. CC BY 4.0. https://doi.org/10.3390/microorganisms12030516












