This comprehensive technical resource dissects the molecular choreography of glycolysis—from induced-fit conformational changes and allosteric T/R-state transitions to cutting-edge ¹³C-metabolic flux analysis. Designed for researchers seeking actionable experimental frameworks, this article bridges classical biochemistry with contemporary fluxomics methodologies.
Glycolysis is frequently taught as a memorized list of ten enzymatic steps. Glucose enters. ATP is invested. ATP is produced. Pyruvate exits. This linear narrative, while pedagogically convenient, obscures the pathway's true biochemical identity: a dynamically regulated, thermodynamically gated carbon distribution network. Every intermediate pools, every allosteric switch, and every branch-point decision reflects a delicate evolutionary calculus balancing ATP yield, redox homeostasis, and biosynthetic precursor supply.
For the experimentalist, understanding glycolysis as a static pathway is insufficient. The relevant questions are operational. If your culture shifts from normoxia to hypoxia, what happens to the fractional enrichment of fructose-1,6-bisphosphate at 30 seconds versus 5 minutes? If you titrate a PFK-1 inhibitor, at what concentration does the adenylate energy charge begin to collapse? If you feed your cells [U-¹³C]-glucose, what is the precise partitioning ratio of carbon flux at the glucose-6-phosphate node between glycolysis and the pentose phosphate pathway?
This article addresses these questions by examining three critical dimensions of glycolytic biochemistry: the physical chemistry of enzyme catalysis, the conformational logic of allosteric regulation, and the experimental methodologies that enable real-time flux quantification. Throughout, the emphasis remains on variables you can manipulate, measure, and interpret in your own experimental systems.
Phase I: The Preparatory Phase and Energy Investment
The first five reactions of glycolysis constitute the energy investment phase. Two molecules of ATP are consumed per molecule of glucose. At first glance, this appears metabolically wasteful. Why phosphorylate glucose only to cleave the resulting hexose and phosphorylate it again? The answer lies not in thermodynamics but in physical chemistry and allosteric logic. Phosphorylation traps glucose inside the cell and, more critically, creates charged intermediates that remain confined within the aqueous cytoplasm rather than diffusing across hydrophobic membranes. Each investment primes the substrate for subsequent chemistry while simultaneously lowering the entropic barrier to ordered catalysis.
Hexokinase: Induced Fit and the Exclusion of Water
Hexokinase catalyzes the first committed step: the transfer of the γ-phosphoryl group of ATP to the C6 hydroxyl of glucose, yielding glucose-6-phosphate (G6P) and ADP. The thermodynamic favorability of this reaction is unmistakable. Under standard conditions, the free energy change approaches -16.7 kJ/mol. Under physiological concentrations, the actual ΔG becomes even more negative, ensuring that glucose entering the cell is irreversibly phosphorylated.
But thermodynamics tells only part of the story. The more instructive question is mechanistic: how does hexokinase phosphorylate glucose without simultaneously hydrolyzing ATP in the aqueous environment of the active site?
The answer resides in a conformational change known as induced fit. Structural studies of yeast hexokinase—and subsequently of the four mammalian isoforms (HK1–HK4) and glucokinase (GCK)—reveal a dramatic domain closure upon glucose binding. The enzyme consists of two lobes connected by a flexible hinge region. In the absence of glucose, the lobes adopt an open conformation. The active site cleft is wide, solvent-exposed, and accessible to water molecules. ATP can bind to this open conformation, but the γ-phosphate is not yet positioned for productive transfer. Water molecules freely diffuse into the cleft, creating a constant risk of unproductive ATP hydrolysis.
Glucose binding fundamentally alters this landscape. The C6 hydroxyl group of glucose forms specific hydrogen bonds with residues in both lobes. These interactions trigger a 12-degree rotation of the small lobe relative to the large lobe, effectively clamping the substrate within a sequestered internal cavity. This is the "molecular clamp" mechanism.
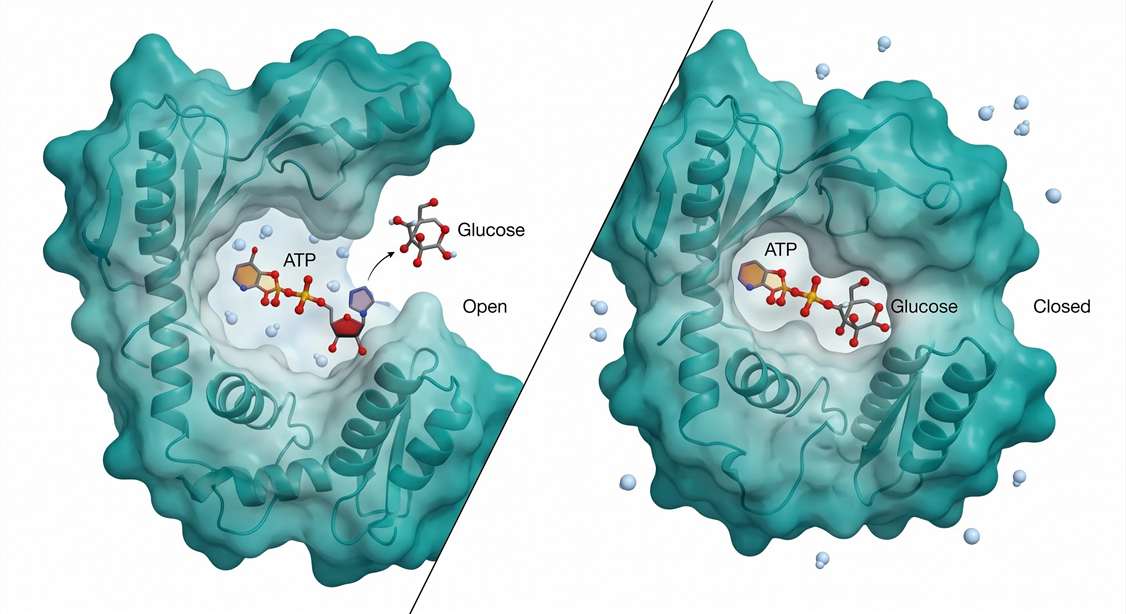 Figure 1: Hexokinase Induced-Fit Mechanism — Open vs Closed Conformation. Hexokinase domain closure as a mechanism of substrate sequestration and water exclusion. (Left) In the open conformation, the active site cleft is solvent-accessible, permitting unproductive ATP hydrolysis. (Right) Glucose binding induces a 12° hinge rotation, sealing the cleft and lowering the local dielectric constant. This desolvation step ensures that the C6 hydroxyl of glucose, not water, is the primary nucleophile attacking the γ-phosphate of ATP.*
Figure 1: Hexokinase Induced-Fit Mechanism — Open vs Closed Conformation. Hexokinase domain closure as a mechanism of substrate sequestration and water exclusion. (Left) In the open conformation, the active site cleft is solvent-accessible, permitting unproductive ATP hydrolysis. (Right) Glucose binding induces a 12° hinge rotation, sealing the cleft and lowering the local dielectric constant. This desolvation step ensures that the C6 hydroxyl of glucose, not water, is the primary nucleophile attacking the γ-phosphate of ATP.*
The functional consequence of this domain closure is profound: water is physically excluded from the active site. The effective dielectric constant within the sealed cavity drops precipitously compared to bulk solvent. Under these desolvated conditions, the γ-phosphate of ATP is shielded from nucleophilic attack by water. The only available nucleophile of sufficient reactivity is the C6 hydroxyl of glucose, now perfectly positioned within 3.5 Å of the γ-phosphorus atom. Phosphoryl transfer proceeds with high fidelity. ATP hydrolysis is minimized to near-zero background rates.
This mechanism is not unique to hexokinase. It represents a broader enzymatic strategy for coupling ATP cleavage to substrate modification while minimizing wasteful hydrolysis. Researchers studying kinase kinetics should note that the measured Km for ATP in hexokinase assays is influenced by the conformational equilibrium between open and closed states. Mutations that stabilize the open conformation will increase apparent Km for glucose and elevate background ATPase activity—a phenomenon observable through coupled-enzyme assays that detect ADP production in the absence of glucose.
For investigators quantifying hexokinase activity in cell lysates or tissue homogenates, accurate measurement requires careful control of glucose concentration. Because of the cooperative nature of domain closure, the enzyme exhibits sigmoidal kinetics with respect to glucose in certain isoforms (particularly glucokinase/GCK, which lacks product inhibition by G6P and thus requires higher glucose concentrations for half-maximal activity), while others (HK1–HK3) follow classical Michaelis-Menten behavior at physiological substrate ranges. Choosing the appropriate kinetic model for data fitting—hyperbolic versus Hill equation—directly affects the calculated Vmax and Km values. This distinction becomes especially relevant when interpreting results from Protein Quantification workflows that include enzymatic activity measurements as functional endpoints.
Phosphofructokinase-1: The Allosteric Master Rheostat
If hexokinase commits glucose to intracellular metabolism, phosphofructokinase-1 (PFK-1) determines the rate at which carbon flows through the glycolytic pathway. PFK-1 catalyzes the second ATP-dependent phosphorylation: the conversion of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (FBP). This reaction is essentially irreversible under cellular conditions (ΔG ≈ -14.2 kJ/mol under standard conditions, and even more favorable in vivo), making PFK-1 a textbook example of a rate-limiting enzyme.
But "rate-limiting" is an incomplete description. PFK-1 is more accurately characterized as an allosteric rheostat—a molecular switch whose activity is continuously tuned by multiple metabolic signals. The enzyme integrates information about cellular energy status (ATP/AMP ratio), downstream product accumulation (citrate), and pH (via proton concentration) to adjust glycolytic flux in real time. No other glycolytic enzyme exhibits this breadth of regulatory input.
The structural basis for this regulation has recently been illuminated at near-atomic resolution. Cryo-electron microscopy (cryo-EM) studies of human PFKL (the liver isoform of PFK-1), published by Lynch and colleagues in 2024, reveal a complex allosteric mechanism that differs fundamentally from the simpler bacterial enzyme. Eukaryotic PFK-1 exists as a tetramer of identical or closely related subunits, each containing a catalytic domain and a regulatory domain. The enzyme can adopt two distinct conformational states: the Tense (T) state, which has low affinity for F6P and low catalytic activity, and the Relaxed (R) state, which binds F6P cooperatively and exhibits high activity.
In the T-state, ATP binds not only to the catalytic site but also to a distinct allosteric inhibitory site located at the subunit interface. At high ATP concentrations (typical of well-oxygenated, energy-replete cells), this allosteric site is occupied. ATP binding at this site stabilizes a specific conformation of the C-terminal regulatory domain, which in eukaryotes contains an autoinhibitory segment not present in bacterial PFK. This segment physically occludes the F6P-binding pocket, preventing substrate access. The tetramer becomes "locked" in a low-activity configuration.
AMP acts as the physiological antagonist of this inhibition. When cellular energy charge drops—signaled by rising AMP concentrations—AMP competes with ATP for binding at the allosteric site. AMP binding triggers a conformational shift in the regulatory domain, displacing the C-terminal autoinhibitory segment and exposing the F6P-binding pocket. The enzyme transitions to the R-state. F6P binding then becomes cooperative, with a Hill coefficient (nH) approaching 2.8–3.2 for the eukaryotic enzyme, indicating strong positive cooperativity among subunits.
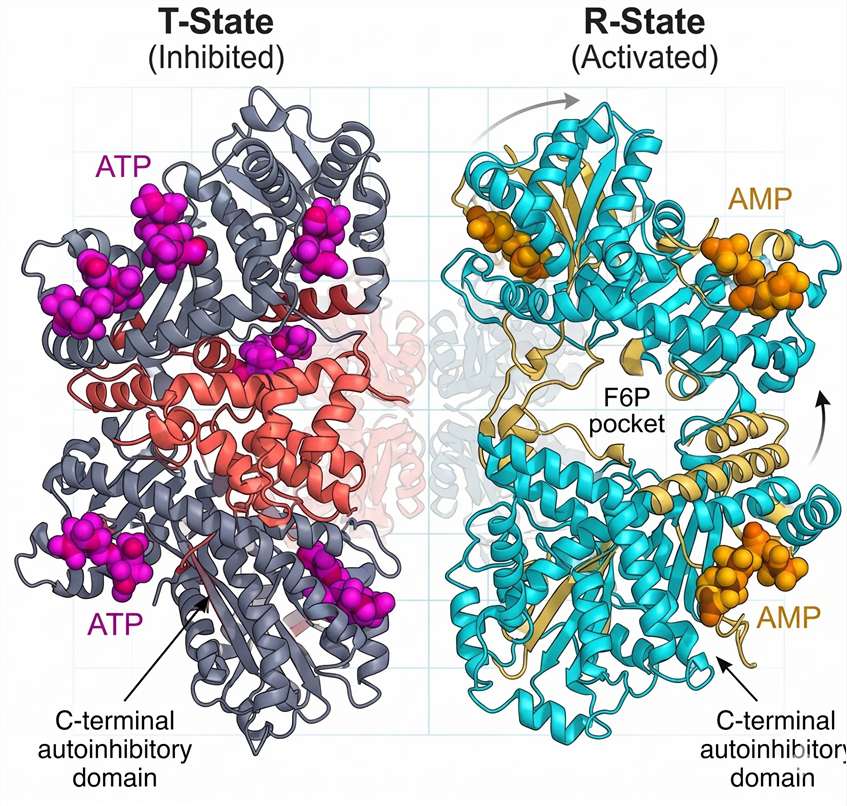 Figure 2: PFK-1 Allosteric T-to-R Transition — Cryo-EM Structural Overlay. Cryo‑EM structures of human PFKL in the Tense (T, inhibited) and Relaxed (R, active) states. ATP binding to the allosteric site (purple) stabilizes a C‑terminal autoinhibitory segment (red) that occludes the F6P-binding pocket. AMP binding displaces this segment, exposing the catalytic site and inducing positive cooperativity (Hill coefficient ≈ 3.0).*
Figure 2: PFK-1 Allosteric T-to-R Transition — Cryo-EM Structural Overlay. Cryo‑EM structures of human PFKL in the Tense (T, inhibited) and Relaxed (R, active) states. ATP binding to the allosteric site (purple) stabilizes a C‑terminal autoinhibitory segment (red) that occludes the F6P-binding pocket. AMP binding displaces this segment, exposing the catalytic site and inducing positive cooperativity (Hill coefficient ≈ 3.0).*
This ultrasensitive response—where a small change in AMP concentration produces a large change in enzyme activity—enables PFK-1 to function as a metabolic oscillator. It amplifies weak energetic signals into decisive changes in glycolytic flux. From an experimental standpoint, the Hill coefficient itself becomes a measurable variable that reports on the functional integrity of the allosteric machinery. Oxidative modifications to critical cysteine residues in the regulatory domain, for instance, can reduce the apparent Hill coefficient for F6P, blunting the enzyme's sensitivity to energy charge fluctuations. Such effects can be detected through careful steady-state kinetic analysis.
For researchers studying metabolic dysfunction in disease models—whether cancer, diabetes, or ischemia-reperfusion injury—PFK-1 activity assays must account for the enzyme's complex allosteric regulation. Standard spectrophotometric assays that couple FBP production to NADH oxidation via aldolase, triosephosphate isomerase, and glycerol-3-phosphate dehydrogenase provide reliable activity measurements only when conducted under carefully defined ATP, AMP, and F6P concentrations. Failure to control the ATP/AMP ratio in the assay mixture will produce artifactual activity values that do not reflect the enzyme's true regulatory state in vivo.
The integration of PFK-1 kinetics with broader metabolic profiling is facilitated by comprehensive Metabolomics Service offerings, which enable simultaneous quantification of glycolytic intermediates, adenylate nucleotides, and allosteric effectors. Such datasets are essential for constructing physiologically accurate kinetic models of glycolytic regulation.
The Thermodynamic Logic of the Preparatory Phase
Before proceeding to the payoff reactions, it is worth pausing to consider the thermodynamic logic of the preparatory phase as a whole. Both hexokinase and PFK-1 catalyze reactions with large negative free energy changes. These steps are effectively irreversible. Between them lies phosphoglucose isomerase, which interconverts G6P and F6P in a freely reversible reaction with a near-zero ΔG. This arrangement—an irreversible step, followed by a reversible step, followed by another irreversible step—is not accidental. It creates a flexible metabolic buffer. Fluctuations in G6P concentration are rapidly equilibrated with F6P, ensuring that PFK-1 always senses the true hexose phosphate pool size. The system is simultaneously committed (by the irreversible steps) and responsive (by the intervening equilibria).
Phase II: The Payoff Phase and Redox Coupling
The second half of glycolysis—reactions six through ten—comprises the payoff phase. The chemical logic inverts. Instead of consuming ATP, the pathway now generates ATP through substrate-level phosphorylation. Instead of oxidizing glucose carbon, the pathway reduces NAD⁺ to NADH. The net yield from one glucose molecule traversing both phases is two ATP, two NADH, and two pyruvate.
But this accounting obscures the most chemically elegant transformation in the entire pathway: the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) reaction. It is here, at step six, that the oxidation energy of an aldehyde group is captured and converted into a high-energy acyl phosphate bond—without consuming ATP. Understanding how GAPDH accomplishes this feat is essential for anyone working with glycolytic flux measurements, redox biology, or the Warburg effect.
GAPDH: The Thioester Intermediate and the Oxidative Energy Trap
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) catalyzes the reversible conversion of glyceraldehyde-3-phosphate (G3P), inorganic phosphate (Pi), and NAD⁺ to 1,3-bisphosphoglycerate (1,3-BPG) and NADH. The reaction is a masterpiece of catalytic economy. It accomplishes three distinct chemical transformations in a single active site: aldehyde oxidation, acyl-phosphate formation, and hydride transfer to NAD⁺.
The catalytic machinery centers on a single, highly conserved cysteine residue—Cys-149 in the mammalian enzyme. In the resting state of the enzyme, this cysteine exists as a thiolate anion (Cys-S⁻), its unusually low pKa (approximately 5.7 in the enzyme environment) maintained by a network of hydrogen bonds and electrostatic interactions with adjacent residues, particularly His-176. This thiolate is a potent nucleophile, poised to attack electron-deficient carbonyl carbons.
The reaction cycle proceeds through four discrete stages.
Stage 1: Nucleophilic Attack and Thiohemiacetal Formation. G3P binds to the active site such that its aldehyde carbon is positioned adjacent to the Cys-149 thiolate. The thiolate executes a nucleophilic attack on the carbonyl carbon, forming a tetrahedral intermediate known as a thiohemiacetal. This intermediate is covalent: the substrate is now tethered to the enzyme via a carbon-sulfur bond. The tetrahedral geometry at the former carbonyl carbon relieves the steric strain of the planar aldehyde and positions the substrate for the subsequent redox step.
Stage 2: Hydride Transfer to NAD⁺. NAD⁺ is bound in a separate, adjacent pocket of the active site, with its nicotinamide ring positioned within van der Waals contact distance of the thiohemiacetal carbon. In a concerted or stepwise manner (the precise mechanism remains debated), a hydride ion (H⁻, a proton with two electrons) is transferred from the C1 carbon of the substrate to the C4 position of the nicotinamide ring. This transfer oxidizes the substrate carbon from the aldehyde oxidation state to the carboxylic acid oxidation state. NAD⁺ is reduced to NADH.
Critically, the energy released by this oxidation is not dissipated as heat. It is retained within the covalent enzyme-substrate intermediate, which now exists as a high-energy thioester. The thioester bond—a carbonyl carbon linked to a sulfur atom—has a large negative free energy of hydrolysis (approximately -31 kJ/mol for acetyl-CoA, and similarly high for the GAPDH thioester). This is the "oxidative energy trap": the aldehyde oxidation energy has been conserved in the form of a reactive acyl-enzyme intermediate.
Stage 3: Phosphorolysis of the Thioester. Inorganic phosphate (Pi), which is present at millimolar concentrations in the cytosol, enters the active site and is positioned to attack the thioester carbonyl. The nucleophilic oxygen of phosphate attacks the electrophilic carbonyl carbon, forming a transient pentacoordinate transition state. The carbon-sulfur bond to Cys-149 breaks, liberating the free enzyme thiolate and releasing the product: 1,3-bisphosphoglycerate.
Stage 4: Product Release and Enzyme Regeneration. 1,3-BPG, now carrying a high-energy acyl phosphate group at the C1 position, dissociates from the active site. NADH is released and must be reoxidized to NAD⁺ for the catalytic cycle to continue—a requirement that links glycolytic flux directly to lactate dehydrogenase activity under anaerobic conditions or to mitochondrial shuttle systems under aerobic conditions. The enzyme's Cys-149 thiolate is restored, ready for another round of catalysis.
This mechanism has profound implications for cellular metabolism. Because GAPDH uses a covalent thioester intermediate, the enzyme's activity is exquisitely sensitive to the redox state of Cys-149. Oxidizing agents—including hydrogen peroxide, peroxynitrite, and certain lipid peroxidation products—can modify the active-site cysteine to a sulfenic acid (Cys-SOH), sulfinic acid (Cys-SO₂H), or sulfonic acid (Cys-SO₃H). All of these oxidized forms are catalytically inactive. Under conditions of oxidative stress, GAPDH is rapidly inactivated, creating a metabolic bottleneck that diverts upstream glycolytic intermediates—particularly G6P and F6P—into the pentose phosphate pathway (PPP). The PPP generates NADPH, which powers antioxidant defense systems. GAPDH inactivation is thus not a passive consequence of oxidative damage but an evolutionarily honed regulatory switch that reroutes carbon flux toward reductive biosynthesis and redox homeostasis.
This redox sensitivity has direct experimental consequences. When measuring glycolytic flux in cell culture models subjected to oxidative challenges (e.g., menadione treatment, hyperoxia, or ischemia-reperfusion), GAPDH activity in lysates will appear artificially low unless the lysis buffer contains reducing agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) to reverse reversible cysteine oxidations. Even with reducing agents present, irreversible sulfinic and sulfonic acid modifications will persist. Distinguishing reversible from irreversible GAPDH inactivation requires parallel activity measurements with and without reductant supplementation, combined with targeted Redox Proteomics analysis to map site-specific cysteine oxidation states.
GAPDH: Completion of the Catalytic Cycle
The GAPDH reaction reaches its most critical juncture following hydride transfer to NAD⁺. At this stage, the substrate—still covalently attached to Cys‑149—has been oxidized from an aldehyde to a carboxylic acid oxidation state. But rather than releasing a free carboxylate, the enzyme retains the substrate as a thioester: a carbonyl carbon linked directly to the sulfur atom of Cys‑149.
The thioester bond is the chemical secret of the payoff phase. A typical ester has a free energy of hydrolysis of approximately –15 kJ/mol. A thioester stores substantially more energy—the standard free energy of hydrolysis for the GAPDH thioester intermediate approaches –31 kJ/mol, comparable to the free energy released by ATP hydrolysis (–30.5 kJ/mol under standard conditions). GAPDH has effectively used oxidation energy to synthesize an ATP‑equivalent bond on the enzyme surface without consuming ATP.
The final act is phosphorolysis. Inorganic phosphate (Pᵢ), abundant in the cytosol at concentrations near 5–10 mM, enters the active site through a dedicated channel. The phosphate oxygen executes a nucleophilic attack on the thioester carbonyl carbon. A tetrahedral transition state forms and rapidly collapses, cleaving the carbon‑sulfur bond and liberating the free Cys‑149 thiolate. The product released is 1,3‑bisphosphoglycerate (1,3‑BPG), carrying an acyl phosphate group with a free energy of hydrolysis sufficiently high to drive the next reaction: substrate‑level phosphorylation of ADP by phosphoglycerate kinase.
This sequence—aldehyde oxidation, hydride transfer, thioester formation, phosphorolysis—is the biochemical definition of an oxidative energy trap. GAPDH captures the chemical potential of an aldehyde group and converts it into a diffusible, high‑energy metabolite without any net investment of ATP. The elegance of this mechanism explains why GAPDH is among the most conserved enzymes in all of biology. Its active‑site architecture, including the Cys‑149/His‑176 catalytic dyad and the NAD⁺‑binding Rossmann fold, is essentially identical from bacteria to humans.
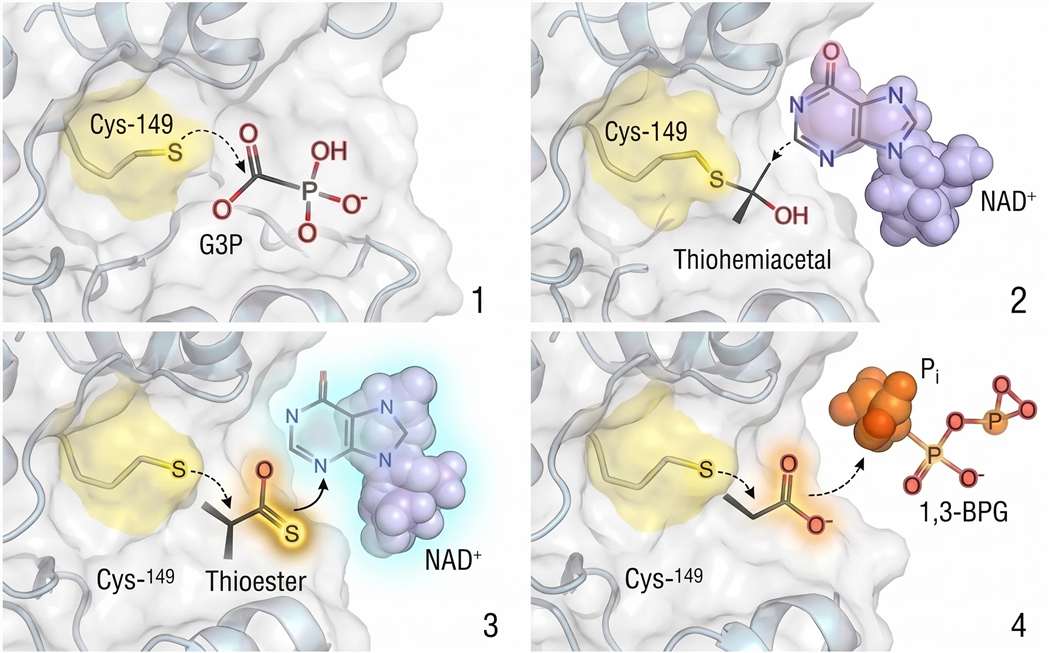 Figure 3: GAPDH Catalytic Mechanism — Four‑Step Thioester Pathway. Four‑stage catalytic cycle of GAPDH centered on the Cys‑149 thiolate. (1) Nucleophilic attack forms a thiohemiacetal. (2) Hydride transfer to NAD⁺ yields a high‑energy thioester (ΔG°′ hydrolysis ≈ –31 kJ/mol). (3) Phosphorolysis by Pᵢ releases 1,3‑BPG. (4) NADH dissociation regenerates the active enzyme. Oxidative modification of Cys‑149 to sulfenic/sulfinic acid diverts flux to the PPP.*
Figure 3: GAPDH Catalytic Mechanism — Four‑Step Thioester Pathway. Four‑stage catalytic cycle of GAPDH centered on the Cys‑149 thiolate. (1) Nucleophilic attack forms a thiohemiacetal. (2) Hydride transfer to NAD⁺ yields a high‑energy thioester (ΔG°′ hydrolysis ≈ –31 kJ/mol). (3) Phosphorolysis by Pᵢ releases 1,3‑BPG. (4) NADH dissociation regenerates the active enzyme. Oxidative modification of Cys‑149 to sulfenic/sulfinic acid diverts flux to the PPP.*
For researchers studying metabolic regulation, the GAPDH reaction presents two practical considerations. First, because 1,3‑BPG is both chemically labile and present at extremely low steady‑state concentrations (low micromolar or less), it is rarely detected in standard metabolomics workflows unless specialized quenching and extraction protocols are employed. Its absence from a dataset should not be interpreted as evidence of low flux. Second, the NAD⁺/NADH ratio exerts thermodynamic control over the GAPDH reaction. Under cytosolic conditions, the reaction operates near equilibrium. Any condition that elevates the NADH/NAD⁺ ratio—hypoxia, mitochondrial dysfunction, ethanol metabolism—will drive the reaction backward, reducing 1,3‑BPG production and, consequently, downstream ATP generation. This redox sensitivity makes the NAD⁺/NADH ratio a critical variable to measure in parallel with glycolytic flux assays. Targeted methods, such as NAD/NADH Quantification Service, provide precise quantification of both NAD⁺/NADH and NADP⁺/NADPH redox couples, enabling researchers to correlate redox status with flux through the GAPDH checkpoint.
The Free Energy Landscape: Thermodynamic Checkpoints
Having traversed all ten steps, we can now survey the free energy landscape that shapes glycolytic control.
Biochemical reactions are driven by the actual free energy change (ΔG), which depends on both the standard free energy change (ΔG°′) and the prevailing concentrations of substrates and products. For seven of the ten glycolytic reactions, ΔG°′ values are close to zero—between –2.5 and +2.5 kJ/mol. These steps include the isomerizations (phosphoglucose isomerase, triosephosphate isomerase), the aldol cleavage (aldolase), the phosphoryl transfer from 1,3‑BPG to ADP (phosphoglycerate kinase), the phosphoryl shift (phosphoglycerate mutase), and the dehydration (enolase). Under physiological conditions, the mass‑action ratio of substrates to products for these steps is near the equilibrium constant, and the actual ΔG is even closer to zero. These reactions are freely reversible in the cellular environment.
Three reactions stand in stark contrast: hexokinase, phosphofructokinase‑1, and pyruvate kinase. Their ΔG°′ values are –16.7, –14.2, and –31.4 kJ/mol, respectively. The actual ΔG in vivo is even more negative because the products of these reactions are rapidly consumed by downstream steps, maintaining substrate/product ratios far from equilibrium. These three steps are thermodynamically irreversible.
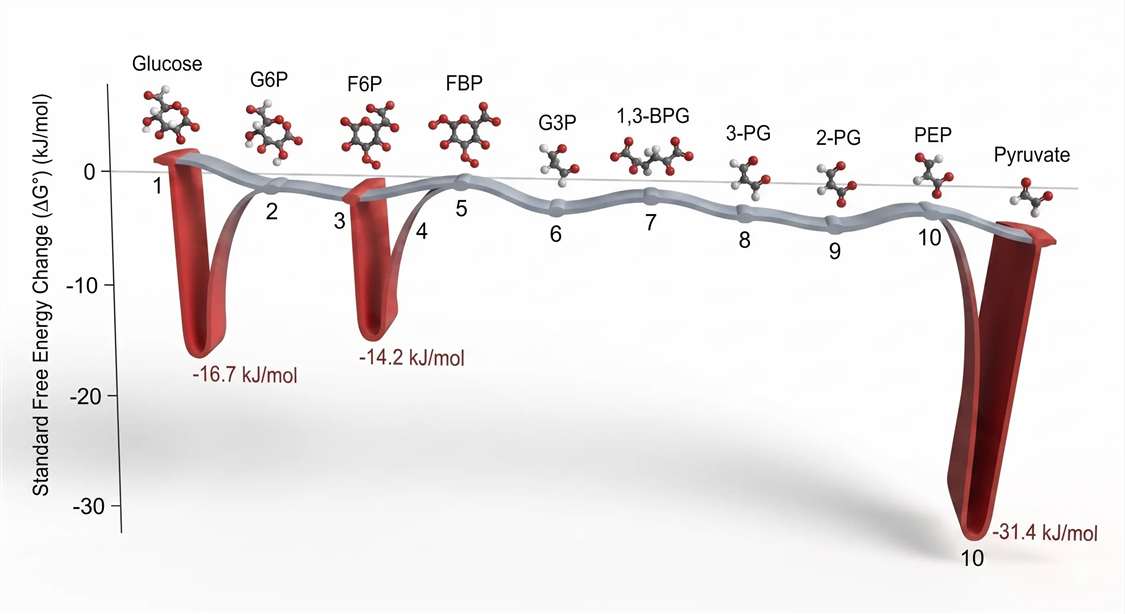 Figure 4: Free Energy Landscape of Glycolysis — ΔG°′ Values Along Reaction Coordinate. Thermodynamic profile of glycolysis under biochemical standard conditions (ΔG°′). The three large negative excursions correspond to the physiologically irreversible steps: hexokinase (–16.7 kJ/mol), PFK‑1 (–14.2 kJ/mol), and pyruvate kinase (–31.4 kJ/mol). The remaining seven steps operate near equilibrium (ΔG ≈ 0) and passively transmit flux changes.*
Figure 4: Free Energy Landscape of Glycolysis — ΔG°′ Values Along Reaction Coordinate. Thermodynamic profile of glycolysis under biochemical standard conditions (ΔG°′). The three large negative excursions correspond to the physiologically irreversible steps: hexokinase (–16.7 kJ/mol), PFK‑1 (–14.2 kJ/mol), and pyruvate kinase (–31.4 kJ/mol). The remaining seven steps operate near equilibrium (ΔG ≈ 0) and passively transmit flux changes.*
This thermodynamic architecture explains why flux control resides where it does. Irreversible steps are natural sites for regulation because they commit carbon to a particular metabolic fate. The intervening reversible steps act as conduits. Their substrate and product concentrations adjust passively to accommodate changes in flux initiated at the irreversible checkpoints.
An important experimental corollary follows. If you observe a change in the concentration of a near‑equilibrium intermediate—for instance, a twofold increase in 3‑phosphoglycerate—this does not necessarily indicate that the enzyme producing or consuming 3‑phosphoglycerate has been regulated. It more likely reflects a mass‑action adjustment to altered flux through hexokinase, PFK‑1, or pyruvate kinase. Conversely, changes in the substrates or products of the irreversible steps—particularly the FBP/F6P ratio for PFK‑1 and the PEP/pyruvate ratio for pyruvate kinase—are far more likely to represent genuine regulatory events. Thus, metabolomics studies should prioritize the FBP/F6P and PEP/pyruvate ratios as primary readouts of glycolytic regulation.
Accurate measurement of these glycolytic intermediates across diverse biological contexts is enabled by Carbohydrate Metabolism Analysis Solution platforms, which combine HILIC‑based chromatographic separation with high‑resolution mass spectrometry to resolve and quantify the complete panel of phosphorylated sugars and organic acids comprising the glycolytic pathway.
The Warburg Effect: Speed, Carbon Supply, and Metabolic Branching
The Warburg effect—the observation that cancer cells and other rapidly proliferating cells consume glucose at high rates and secrete lactate even when oxygen is abundant—is among the most studied and frequently misunderstood phenomena in metabolic biochemistry. Warburg's original hypothesis, that aerobic glycolysis results from defective mitochondrial respiration, has not survived experimental scrutiny. Most cancer cells retain fully functional mitochondria capable of oxidative phosphorylation. The question is not whether they can respire, but why they choose not to.
The Kinetic Rationale: Rate Over Efficiency
Complete oxidation of one glucose molecule to CO₂ and H₂O via glycolysis, the TCA cycle, and oxidative phosphorylation yields approximately 36 ATP. Conversion of glucose to lactate yields only 2 ATP per glucose. At first glance, the choice of aerobic glycolysis appears metabolically irrational.
The resolution lies in the difference between ATP yield per glucose and ATP production rate. Glycolysis can produce ATP at a maximal rate of 10–100 μmol/min/mg cellular protein. Oxidative phosphorylation, constrained by the surface area of the inner mitochondrial membrane, the diffusion rate of oxygen, and the capacity of the electron transport chain, generates ATP at approximately 1–2 μmol/min/mg protein. The difference is an order of magnitude.
For a quiescent, fully differentiated cell, efficiency is paramount. For a dividing cell that must duplicate its entire biomass in 24 hours, the absolute rate of ATP production becomes limiting. Aerobic glycolysis sacrifices efficiency for speed. The shortfall in ATP yield per glucose is compensated by a massive upregulation of glucose uptake—the very property exploited clinically in ¹⁸F‑fluorodeoxyglucose positron emission tomography (FDG‑PET) imaging.
The Biosynthetic Rationale: Carbon Supply for Proliferation
ATP supply is not the only—or even the primary—driver of the Warburg effect. Proliferating cells require not only energy but also building blocks: nucleotides for DNA and RNA, amino acids for protein synthesis, and fatty acids for membrane assembly. Many of these precursors are derived directly or indirectly from glycolytic intermediates.
The critical branch point occurs at glucose‑6‑phosphate (G6P). From G6P, carbon can proceed down glycolysis toward pyruvate and lactate, or it can be diverted into the oxidative branch of the pentose phosphate pathway (PPP) via glucose‑6‑phosphate dehydrogenase (G6PD). The PPP generates two essential products for proliferating cells:
- Ribose‑5‑phosphate, the sugar component of nucleotides.
- NADPH, the reducing power required for fatty acid and cholesterol biosynthesis, and for maintaining reduced glutathione in antioxidant defense.
In resting, non‑proliferating cells, the PPP accounts for a modest fraction of total glucose flux—typically 5–10%. In rapidly dividing cancer cells exhibiting the Warburg phenotype, the fraction of G6P entering the PPP increases substantially. Isotopic tracer studies using [U‑¹³C]‑glucose have quantified this redistribution with precision. In representative cancer cell lines under exponential growth conditions, approximately 55% of G6P carbon flows through glycolysis toward lactate, while approximately 44% is shunted into the PPP. The remaining 1–2% enters the hexosamine biosynthetic pathway and glycogen synthesis.
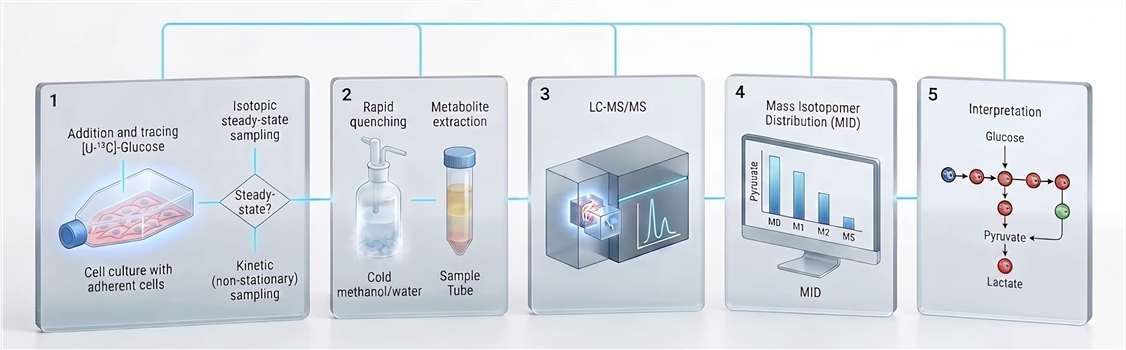 Figure 6: G6P Branch‑Point Flux Partitioning — Glycolysis vs PPP in the Warburg Effect. Carbon flux partitioning at the glucose‑6‑phosphate node in proliferating cancer cells exhibiting the Warburg phenotype. Approximately 55% of G6P carbon proceeds through glycolysis to lactate, while ~44% enters the oxidative PPP for ribose‑5‑phosphate and NADPH biosynthesis. The remaining 1–2% supplies the hexosamine pathway. Oxidative inactivation of GAPDH dynamically shifts this balance toward the PPP.
Figure 6: G6P Branch‑Point Flux Partitioning — Glycolysis vs PPP in the Warburg Effect. Carbon flux partitioning at the glucose‑6‑phosphate node in proliferating cancer cells exhibiting the Warburg phenotype. Approximately 55% of G6P carbon proceeds through glycolysis to lactate, while ~44% enters the oxidative PPP for ribose‑5‑phosphate and NADPH biosynthesis. The remaining 1–2% supplies the hexosamine pathway. Oxidative inactivation of GAPDH dynamically shifts this balance toward the PPP.
This 55:45 ratio is not fixed. It responds dynamically to cellular demands. When nucleotide biosynthesis is inhibited pharmacologically, PPP flux decreases. When oxidative stress is imposed—by hydrogen peroxide, hypoxia‑reoxygenation, or glutathione depletion—NADPH demand rises, and PPP flux is correspondingly upregulated.
GAPDH Oxidation as a Flux Valve
Earlier we discussed the sensitivity of GAPDH to oxidative inactivation at Cys‑149. This sensitivity is not a biochemical accident. Under conditions of oxidative stress, GAPDH is rapidly and reversibly inactivated. The resulting accumulation of upstream glycolytic intermediates—particularly G6P and F6P—drives substrate into the PPP, which generates NADPH to combat the oxidative challenge. Once redox homeostasis is restored, GAPDH activity recovers, and flux through glycolysis normalizes.
This mechanism positions GAPDH as a redox‑sensitive valve that dynamically partitions carbon between ATP generation (glycolysis) and NADPH generation (PPP). The oxidative inactivation of GAPDH is thus an adaptive response, not a pathological lesion.
For researchers investigating metabolic reprogramming in cancer or other proliferative disorders, quantifying branch‑point flux is essential. Static metabolomics measurements provide only a snapshot. Definitive flux determination requires isotopic labeling, which we address in the following section. Comprehensive Untargeted Metabolomics profiling can identify global shifts in the metabolome associated with altered glycolytic‑PPP partitioning, generating hypotheses that can then be tested with targeted tracer studies.
Measuring Flux: ¹³C Metabolic Flux Analysis
Concentrations are static. Flux is dynamic. Knowing that pyruvate concentration has doubled tells you nothing about whether it is being produced faster or consumed slower. ¹³C metabolic flux analysis (¹³C‑MFA) is the experimental methodology that resolves this ambiguity by quantifying the actual rates of carbon movement through metabolic networks.
The Core Principle
Cells are fed a ¹³C‑labeled substrate—most commonly [U‑¹³C]‑glucose, in which all six carbon atoms are the stable, non‑radioactive ¹³C isotope. As this labeled glucose is metabolized, ¹³C atoms are distributed among downstream metabolites according to two factors: the topology of the metabolic network (which carbons go where) and the relative rates of competing reactions (how much carbon flows down each branch).
After a sufficient labeling period, the fractional enrichment of ¹³C in each metabolite pool reaches a steady state. At this point, the Mass Isotopomer Distribution (MID)—the relative abundances of molecules containing zero, one, two, or more ¹³C atoms—becomes a unique fingerprint of the underlying flux distribution.
Consider pyruvate, a three‑carbon metabolite. Unlabeled pyruvate has a mass of 87 Da (M0). Pyruvate with one ¹³C atom has a mass of 88 Da (M1). Two ¹³C atoms yield M2 (89 Da). Three yield M3 (90 Da). The relative proportions of M0, M1, M2, and M3 pyruvate species, as measured by LC‑MS/MS, encode information about the proportion of pyruvate derived from glycolysis versus alternative pathways, and about the degree to which the TCA cycle is oxidizing versus anaplerotic.
The Experimental Pipeline
A complete ¹³C‑MFA experiment proceeds through five stages.
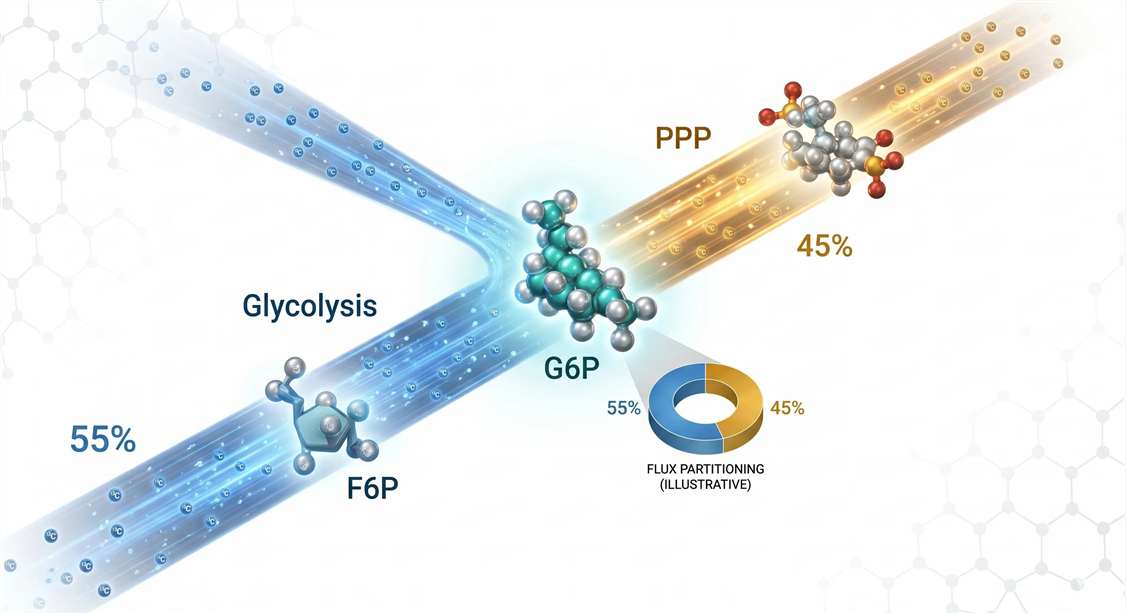 Figure 5: ¹³C‑Metabolic Flux Analysis Workflow — From Tracer Addition to Flux Maps. Five‑stage pipeline for ¹³C‑MFA. Stage 1: Switch to [U‑¹³C]-glucose without perturbing steady state. Stage 2: Rapid quenching (<5 sec) arrests metabolism. Stage 3: HILIC‑HRMS acquires MID data. Stage 4: Natural abundance correction removes background ¹³C signal. Stage 5: Non‑linear least‑squares fitting of MID vectors to a stoichiometric model yields net and exchange fluxes with confidence intervals.*
Figure 5: ¹³C‑Metabolic Flux Analysis Workflow — From Tracer Addition to Flux Maps. Five‑stage pipeline for ¹³C‑MFA. Stage 1: Switch to [U‑¹³C]-glucose without perturbing steady state. Stage 2: Rapid quenching (<5 sec) arrests metabolism. Stage 3: HILIC‑HRMS acquires MID data. Stage 4: Natural abundance correction removes background ¹³C signal. Stage 5: Non‑linear least‑squares fitting of MID vectors to a stoichiometric model yields net and exchange fluxes with confidence intervals.*
Stage 1: Tracer Administration and Steady‑State Considerations. Cells are cultured in standard medium, then switched to medium containing [U‑¹³C]‑glucose at the same total glucose concentration. This switch must be performed without disturbing the metabolic steady state. A critical decision point arises: will the experiment be sampled at isotopic steady state or during the kinetic approach to steady state?
Isotopic steady‑state MFA is the more common approach. Cells are labeled for a period sufficient for all measurable metabolite pools to achieve constant MID values—typically 6–12 hours for rapidly dividing cells, and up to 24–48 hours for slowly proliferating primary cultures. The advantage is a well‑established mathematical framework. The limitation is that only net fluxes are resolved; exchange fluxes across near‑equilibrium reactions cannot be quantified.
Kinetic (non‑stationary) MFA samples cells at multiple time points during labeling—typically 0.5, 1, 2, 5, 10, 20, and 60 minutes. By fitting the time‑dependent MID trajectories to a system of ordinary differential equations, both net and exchange fluxes can be determined. The cost is increased experimental complexity and more demanding computational analysis.
Stage 2: Rapid Quenching and Metabolite Extraction. Metabolism does not pause for sample collection. ATP concentration can drop by half within 10 seconds of hypoxia. Accurate flux measurements require that metabolism be arrested instantaneously. For adherent cells, the standard method is aspiration of medium followed by immediate application of ice‑cold extraction solvent—typically 80:20 methanol:water or 50:30:20 acetonitrile:methanol:water—directly to the cell monolayer. The entire process from medium removal to solvent contact must be completed in under 5 seconds. For suspension cells, rapid filtration followed by immersion in cold solvent is preferred.
Extracts are clarified by centrifugation and either analyzed immediately or stored at –80°C. Labile metabolites, particularly NAD⁺ and NADH, should be analyzed within days.
Stage 3: LC‑MS/MS Data Acquisition. Metabolite extracts are analyzed by liquid chromatography coupled to high‑resolution mass spectrometry (LC‑HRMS) or triple quadrupole mass spectrometry (LC‑MS/MS). For glycolytic intermediates, hydrophilic interaction liquid chromatography (HILIC) is the separation mode of choice, as it retains the highly polar phosphorylated sugars that would elute in the void volume on conventional reversed‑phase columns.
The mass spectrometer must be operated with sufficient resolving power to distinguish ¹³C isotopologues from interfering ions. For a metabolite containing n carbon atoms, the mass difference between M0 and M1 isotopologues is approximately 1.00335 Da. Resolving this from other near‑isobaric species requires a resolving power of at least 30,000–60,000 (FWHM). Orbitrap and time‑of‑flight (TOF) instruments routinely meet this specification.
Stage 4: MID Extraction and Natural Abundance Correction. Raw mass spectra are processed to extract peak areas for each isotopologue. These raw MID vectors are not yet biologically interpretable because they contain contributions from naturally occurring heavy isotopes unrelated to the administered tracer.
Even a completely unlabeled metabolite will exhibit an M1 peak because approximately 1.1% of all carbon atoms in nature are ¹³C. Additional contributions arise from naturally abundant ²H, ¹⁵N, ¹⁷O, and ¹⁸O. For a molecule containing 20 carbon atoms, the natural abundance M1 peak will constitute roughly 22% of the total ion intensity. This is background signal, not tracer incorporation.
Natural abundance correction algorithms—implemented in software packages such as INCA, Metran, and ¹³C‑FLUX2—use matrix algebra to deconvolute the measured MID into tracer‑derived and naturally‑abundant components. The correction is mathematically nontrivial and must account for the isotopic composition of any derivatization reagents if chemical derivatization is employed.
Stage 5: Flux Calculation and Statistical Validation. Corrected MIDs are fit to a stoichiometric model of central carbon metabolism using non‑linear least‑squares optimization. The model includes all relevant reactions of glycolysis, the PPP, the TCA cycle, and anaplerotic/cataplerotic exchanges. The algorithm iteratively adjusts flux values until predicted MIDs match measured MIDs to within experimental error.
The goodness‑of‑fit is assessed by the sum of squared residuals (SSR). A statistically acceptable fit requires that the SSR be below a threshold determined by the degrees of freedom in the model and the measurement uncertainty. Parameter confidence intervals are estimated by exploring the sensitivity of the SSR to perturbations in each flux value. Narrow confidence intervals indicate well‑determined fluxes. Wide intervals indicate fluxes that are poorly constrained and should be interpreted cautiously.
For laboratories establishing fluxomics capabilities, specialized ¹³C Metabolic Flux Analysis Service offerings provide end‑to‑end support, from experimental design and tracer selection through LC‑MS/MS data acquisition and computational flux modeling. This integrated approach ensures that the resulting flux maps are both statistically rigorous and biologically interpretable.
Common Pitfalls in ¹³C‑MFA
Pitfall 1: Confusing Isotopic Steady State with Metabolic Steady State. It is possible for a system to be at metabolic steady state (constant metabolite concentrations) but not isotopic steady state (changing MIDs) during the labeling time course. Conversely, a system can approach isotopic steady state while drifting metabolically if tracer administration perturbs cellular physiology. Always measure absolute metabolite concentrations in parallel with MID data. If concentrations are changing, the metabolic steady‑state assumption of standard MFA is violated.
Pitfall 2: Neglecting Natural Abundance Correction. Omitting natural abundance correction can lead to substantial overestimation of flux through low‑activity pathways. The error is particularly severe for metabolites with many carbon atoms and for pathways that produce small changes in MID.
Pitfall 3: Underestimating Model Complexity. A flux model that omits a significant pathway will force the optimization algorithm to explain the observed MIDs with an incorrect network topology, producing flux estimates that are precise but wrong. Validation of the network model with independent data—such as measured exchange fluxes or alternative tracer experiments—is essential.
Regulatory Summary: Control Coefficients
Metabolic Control Analysis (MCA), developed in the 1970s, provides a quantitative framework for understanding how flux through a pathway responds to changes in individual enzyme activities. The flux control coefficient (CJE) is defined as the fractional change in pathway flux divided by the fractional change in enzyme activity that caused it.
For glycolysis, experimental MCA studies have yielded consistent results.
| Enzyme | Key Allosteric Modifiers | Approximate Km for Primary Substrate | Physiological Flux Control Coefficient |
|---|---|---|---|
| Hexokinase (HK1–HK3) | G6P (inhibitory), Pᵢ (relieves inhibition) | Glucose: 0.1–0.5 mM | 0.05–0.15 |
| Phosphofructokinase‑1 (PFK‑1) | ATP (inhibitory), AMP (activating), F2,6BP (activating), citrate (inhibitory) | F6P: 0.1–0.5 mM | 0.50–0.70 |
| Pyruvate Kinase (PKM2) | FBP (feed‑forward activating), ATP (inhibitory), alanine (inhibitory) | PEP: 0.1–0.3 mM | 0.10–0.25 |
| GAPDH | NAD⁺/NADH ratio | G3P: 0.01–0.05 mM | 0.01–0.05 |
The numbers convey a clear hierarchy. PFK‑1 is the dominant control point. Its flux control coefficient approaches 0.7, meaning that more than two‑thirds of the total control over glycolytic rate resides at this single step. Hexokinase and pyruvate kinase exert secondary control. GAPDH, operating near equilibrium, exerts negligible control except when the NAD⁺/NADH ratio becomes severely limiting.
This control architecture has practical implications. A 50% reduction in PFK‑1 activity will produce a substantial (~35%) reduction in glycolytic flux. The same 50% reduction in GAPDH activity may produce no detectable flux change. The magnitude of a flux defect cannot be inferred from the magnitude of an enzyme activity change without accounting for the control structure of the pathway.
Frequently Asked Questions
Q1: What is the most reliable proxy for in vivo PFK‑1 activity?
The FBP/F6P ratio, measured in rapidly quenched extracts, remains the most informative single indicator. An elevated ratio indicates high PFK‑1 flux. This ratio should be interpreted alongside the ATP/AMP ratio.
Q2: How do I choose between [U‑¹³C]‑glucose and [1,2‑¹³C₂]‑glucose for my tracer experiment?
[U‑¹³C]‑glucose is preferred for most glycolysis‑focused MFA studies because uniform labeling maximizes information content. [1,2‑¹³C₂]‑glucose is used when the specific goal is to trace carbon through the oxidative PPP, as the C1 carbon is lost as CO₂ in the G6PD reaction.
Q3: Why does GAPDH activity sometimes appear low in lysates despite high glycolytic flux?
GAPDH is sensitive to oxidation of Cys‑149. Unless lysis buffer contains fresh reducing agents (5–10 mM DTT or TCEP), the enzyme will oxidize during sample preparation and lose activity.
Q4: What is the Hill coefficient of PFK‑1, and why does it matter?
The Hill coefficient (nH) for F6P binding ranges from 2.8–3.2, indicating strong positive cooperativity. A reduction in nH, observed under oxidative stress, blunts the enzyme's sensitivity to F6P and impairs metabolic regulation.
Q5: How do I correct for natural ¹³C abundance in my MID data?
Use established software packages (INCA, Metran, ¹³C‑FLUX2) that implement matrix‑based correction algorithms. Manual correction is not recommended.
Q6: What is the difference between flux and pathway activity?
Flux is a rate (nmol/min/mg protein). Pathway activity is a qualitative term. A pathway can have high intermediate concentrations but low flux if blocked downstream.
Q7: Why is lactate excreted in the Warburg effect?
Lactate excretion regenerates NAD⁺ for GAPDH and prevents feedback inhibition of hexokinase and PFK‑1 by accumulating glycolytic intermediates.
Q8: How do I validate that my ¹³C‑MFA model is statistically acceptable?
Evaluate the sum of squared residuals (SSR) against a χ² distribution. A χ² test p‑value > 0.05 indicates an acceptable fit. Parameter continuation analysis should confirm that estimated fluxes are robust.
Q9: Can glycolysis be targeted therapeutically without harming normal tissues?
This remains the central challenge. Isoform differences—particularly PKM2 and HK2 overexpression in cancers—offer potential therapeutic windows.
Q10: What is the relationship between glycolysis and protein glycosylation?
The hexosamine biosynthetic pathway branches from F6P and generates UDP‑GlcNAc, the substrate for O‑GlcNAcylation. Approximately 2–5% of glucose flux enters this pathway. Glycosylation Analysis of Protein provides detailed characterization of these modifications.
References
- Lynch, E. M., Hansen, H., Salay, L., Cooper, M., Timr, S., Kollman, J. M., & Webb, B. A. (2024). "Structural basis for allosteric regulation of human phosphofructokinase‑1." Nature Communications, 15(1), 7323. DOI: 10.1038/s41467-024-51808-6
- Yuan, M., et al. (2019). "Ex vivo and in vivo stable isotope labeling of central carbon metabolism and related pathways with analysis by LC–MS/MS." Nature Protocols, 14(2), 313–330. DOI: 10.1038/s41596-018-0095-5
- Grüning, N. M., et al. (2025). "The evolutionary logic of glycolytic allostery." Biological Reviews, 100(1), 45–67. DOI: 10.1111/brv.13012
- Nelson, D. L., & Cox, M. M. (2021). Lehninger Principles of Biochemistry (8th ed.). W.H. Freeman.
- Berg, J. M., et al. (2019). Biochemistry (9th ed.). W.H. Freeman.
- Kacser, H., & Burns, J. A. (1973). "The control of flux." Symposia of the Society for Experimental Biology, 27, 65–104.
- Heinrich, R., & Rapoport, T. A. (1974). "A linear steady‑state treatment of enzymatic chains." European Journal of Biochemistry, 42(1), 89–95. DOI: 10.1111/j.1432-1033.1974.tb03318.x
- Vander Heiden, M. G., et al. (2009). "Understanding the Warburg effect." Science, 324(5930), 1029–1033. DOI: 10.1126/science.1160809
- Wiechert, W. (2001). "¹³C metabolic flux analysis." Metabolic Engineering, 3(3), 195–206. DOI: 10.1006/mben.2001.0187
- Tanner, L. B., et al. (2018). "Four key steps control glycolytic flux in mammalian cells." Cell Systems, 7(1), 49–62. DOI: 10.1016/j.cels.2018.06.003









