This guide is for researchers who already know the vocabulary of tandem MS but need a mechanism-based way to read why spectra change across precursor forms, activation modes, and analytical contexts.
Mass spectrometry is often taught as a list of names. Precursor ions enter. Product ions appear. CID gives b- and y-ions. ETD gives c- and z-ions. A phosphate loss means one thing. A water loss means another. That model is useful at the beginning, but it stops being enough the moment a spectrum becomes mixed, energy-sensitive, or strongly chemistry-dependent.
A better model starts one step earlier. A tandem spectrum is not just a record of what broke. It is a record of what ion formed, where charge was held, how energy was deposited, and which dissociation routes became accessible before competing pathways took over. Once that sequence is clear, many practical problems become easier to interpret. Why does the same precursor fragment cleanly on one platform and poorly on another? Why does a phosphopeptide collapse into neutral loss under one mode and retain its modification under another? Why can a strong search score still coexist with chemically inconsistent peaks?
The central logic is simple: molecular structure shapes ion formation, ion formation shapes precursor behavior, precursor behavior shapes fragmentation, and fragmentation shapes analytical identification confidence. The classic mobile proton framework remains central to that logic, while electron-based and photon-based activation methods show why not all tandem MS belongs to the same chemical regime.
Ion formation decides more than the precursor m/z
Before a molecule fragments, it must survive ionization as a specific gas-phase ion. That step is often treated as housekeeping. It is not. The precursor already reflects a chemical filter imposed by solvent composition, salt background, desolvation efficiency, proton affinity, cation-binding strength, and source conditions.
That is why one analyte rarely enters tandem MS as one universal species. In electrospray, the same molecule may appear mainly as ([M+H]^+), mainly as ([M+Na]^+), partly as ([M-H]^-), or as a mixture. Those choices are not cosmetic. They reshape the entire downstream fragmentation map.
Protonated, sodiated, and deprotonated ions are different starting chemistries
The protonated ion, ([M+H]^+), is the default mental model for many analysts because it is common and often useful. But protonation succeeds only when the analyte can stabilize the extra proton through a basic or polar site that remains favorable during the transition from solvent droplet to gas phase. A protonated precursor is therefore not just the neutral molecule plus one unit of mass. It is a specific charged structure with a specific charge topology.
Sodiated ions, ([M+Na]^+), follow a different rule set. For oxygen-rich molecules, sodium coordination can compete strongly with protonation. This is why many carbohydrates, glycolipids, and oxygen-dense metabolites continue to show metal adducts even when the analyst would prefer a simpler protonated precursor. Once selected, those adducts do not fragment like protonated ions with a harmless mass shift. Charge localization changes. Conformation changes. Accessible dissociation routes change too.
Negative-mode ions make the same point from the opposite direction. A deprotonated precursor, ([M-H]^-), often becomes more informative for acidic metabolites, phosphates, sulfates, and carboxylate-rich species because the site of deprotonation changes the active cleavage logic. A compound that gives poor structural evidence in positive mode may become highly readable in negative mode because the gas-phase chemistry is no longer the same.
This is why precursor selection should be treated as part of the interpretation strategy, not just part of method setup. In practice, workflows built around high-resolution exact-mass confirmation become much more powerful when they distinguish protonated and metal-adducted precursor behavior instead of collapsing them into one formula-assignment step.
Solvent effects and proton affinity quietly shape the precursor pool
Adduct competition is often explained too loosely. Analysts say that acid helps protonation and residual salts promote metal adduction. Both statements are true, but incomplete. The real competition depends on proton affinity, gas-phase basicity, local coordination geometry, and the kinetics of droplet shrinkage and desolvation.
That is why precursor distributions can change after what looks like a minor method edit. A different sample cleanup step, a different buffer system, or a different residual sodium load can shift the dominant ion form without changing the underlying analyte. What looks like source inconsistency is often just a new thermodynamic competition becoming visible.
The practical lesson is short: do not ask only what the precursor mass is. Ask what ion chemistry produced it.
Isotopic clusters help constrain what the precursor can be
The monoisotopic peak is only the beginning. The isotopic envelope often carries enough information to narrow the elemental space before MS/MS starts. The M+1 region reflects contributions such as (^{13}C). The M+2 region becomes especially useful when sulfur, chlorine, or bromine reshape the cluster in characteristic ways. That is why formula generation in accurate-mass workflows depends on isotopic fit as well as ppm error.
The broader logic behind the "Seven Golden Rules" is precisely this: plausible formulas are filtered not just by mass, but by isotope behavior and chemical plausibility checks. A candidate formula that matches exact mass but fails the isotopic pattern is not a strong answer. It is a mismatch hiding behind low ppm error.
Even-electron and odd-electron ions do not fragment by the same logic
One of the most important divides in mass spectrometry is the split between even-electron and odd-electron ions. Electrospray usually produces closed-shell, even-electron ions. These often fragment through heterolytic and rearrangement-assisted pathways. Much of peptide CID and HCD interpretation assumes this regime.
Radical cations are different. They are odd-electron ions, and radical pathways open homolytic channels that are weak or inaccessible in closed-shell systems. That difference matters far beyond textbook terminology. It changes which bonds are likely to break, which rearrangements become competitive, and how predictable the product-ion family will be under a given activation regime.
This is one reason ETD and ECD are so important. They do not merely fragment "gently." They move multiply charged precursors into radical chemistry, and that changes the cleavage map.
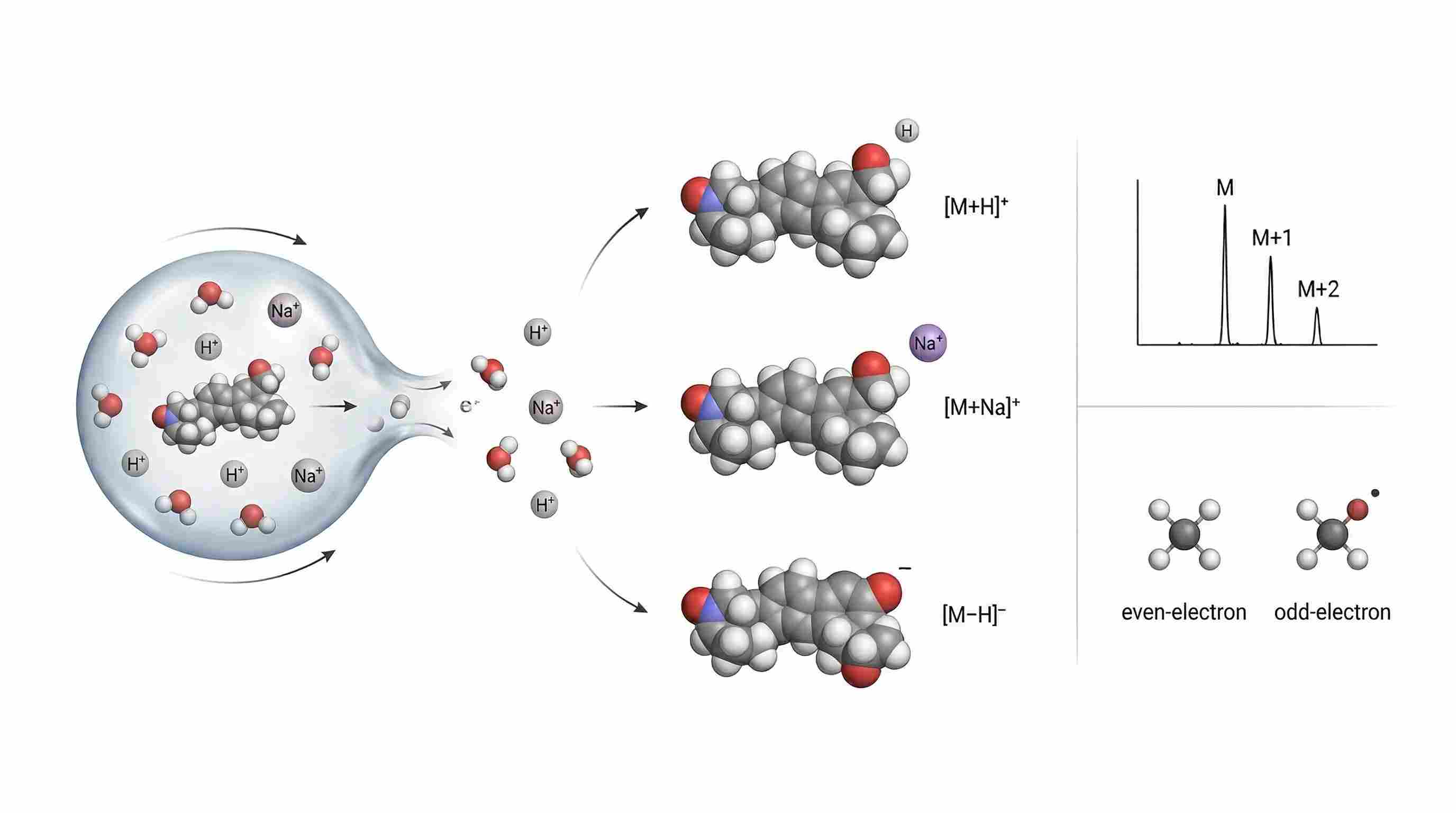 Figure 1. Ion formation, adduct competition, isotopic filtering, and precursor class split. Use one shared analyte and show four chemically distinct precursor outcomes: protonated, sodiated, deprotonated, and radical-like precursor class contrast. Add a compact isotopic-envelope inset with M, M+1, and M+2. The design goal is to show that precursor forms are different chemical starting points, not interchangeable mass labels.
Figure 1. Ion formation, adduct competition, isotopic filtering, and precursor class split. Use one shared analyte and show four chemically distinct precursor outcomes: protonated, sodiated, deprotonated, and radical-like precursor class contrast. Add a compact isotopic-envelope inset with M, M+1, and M+2. The design goal is to show that precursor forms are different chemical starting points, not interchangeable mass labels.
The mobile proton model is still the best entry point into peptide fragmentation
If one concept explains more peptide MS/MS behavior than any other, it is the mobile proton model. Not because it explains everything, but because it explains so many real spectra that would otherwise look arbitrary.
The short form of the model is simple: fragmentation depends strongly on whether a proton can migrate to productive sites along the ion during activation. If a proton is mobile, charge-directed cleavage of the backbone becomes easier. If that proton is trapped at a strongly basic site, productive backbone fragmentation may weaken, shift, or lose to less useful pathways.
That is why collision energy alone does not determine spectrum quality. Two peptide ions can absorb comparable activation and still produce very different fragment ladders because their internal charge topologies are different. Paizs and Suhai's review remains foundational here because it frames peptide fragmentation not as random breakage, but as a hierarchy of chemical channels that the mobile proton model helps rationalize, especially for low-energy CID.
Charge state matters, but proton mobility matters more
Analysts often start with precursor charge state. A doubly charged peptide is expected to behave differently from a triply charged one. That is true, but it is not the deepest variable. What matters more is where the proton or protons are effectively held, and whether one of them can become mobile during activation.
Basic residues shape that outcome. Arginine is especially important because it can sequester a proton strongly. Lysine and histidine also influence the charge map. A peptide that locks protonation onto a highly basic site may show weaker or more biased backbone cleavage because the proton needed to activate the amide system is not easily redistributed. By contrast, when at least one proton remains mobile, charge-directed cleavage becomes much more accessible.
This helps explain a familiar experimental pattern. Some doubly charged peptides give long, interpretable b/y ladders. Others of the same charge state do not. The difference is often not instrument quality. It is local chemistry.
Proton migration is a kinetic event, not just a structural idea
The phrase "mobile proton" sounds static, but it is kinetic. During low-energy collisional activation, internal energy rises through many small collisions. As the ion samples higher-energy conformations, proton transfer among polar and basic sites becomes more feasible. If a proton reaches a productive backbone environment soon enough, cleavage follows. If it remains trapped, the precursor may instead survive longer, lose small neutrals, or fragment through fewer informative channels.
That is why CID should be read as pathway competition under gradual heating, not as uniform breakage. Different channels open at different thresholds. The ones that become accessible first, or remain accessible long enough to dominate, shape the observed spectrum.
The backbone does not cleave uniformly because the activation barrier is local
Peptide bonds are not equally easy to break in the gas phase. Charge-directed cleavage still requires access to an activation pathway, and that pathway is shaped by local structure. Carbonyl participation matters. Neighboring residues matter. Proton availability matters. Conformation matters.
This is why fragmentation ladders are uneven. Some bonds break readily. Others remain weak even when more energy is added. Missing fragments are therefore not always signs of bad data. They often reflect real energetic bias in the precursor.
A useful reading habit follows from that point. When a backbone region is underrepresented, do not assume failure first. Ask what local chemistry made that region harder to access.
A practical readout for real spectra
The mobile proton model becomes most useful when it changes how you inspect a spectrum. Three quick checks help.
First, look at backbone continuity. A long and reasonably coherent b/y ladder often suggests that at least one proton remained sufficiently mobile for productive charge-directed cleavage. Second, look for bias near strongly basic residues. If cleavage appears suppressed or redistributed around arginine-rich regions, proton sequestration may be part of the explanation. Third, ask whether neutral loss is consuming the signal budget. When dominant small-molecule losses crowd out backbone evidence, the issue may be pathway competition rather than poor acquisition.
That is where tandem MS stops being a naming exercise and becomes an evidence-reading exercise. The question is not simply which ions are present. The question is which chemistry won.
In practice, that distinction matters for peptide sequencing workflows based on tandem MS evidence and for de novo sequencing strategies built around incomplete fragment ladders, because both depend on understanding what the missing parts of a ladder actually mean.
Charge-directed and charge-remote fragmentation belong to different worlds
The mobile proton model is most at home in peptide analysis, but its value becomes even clearer when contrasted with charge-remote fragmentation. In charge-directed fragmentation, the charge site actively helps open the cleavage route. Proton mobility lowers barriers and guides bond rupture. This is the core logic behind much peptide CID and HCD behavior.
In charge-remote fragmentation, cleavage can occur at positions that are structurally distant from the formal charge site. That becomes especially important in lipid analysis, where hydrocarbon-chain fragmentation, headgroup behavior, and unsaturation-sensitive channels do not always depend on peptide-like proton transfer. The activation label on the instrument may still say CID or HCD, but the mechanistic meaning is different because the role of charge is different.
That is why peptide fragmentation rules should not be copied directly into lipidomics or metabolomics. The analyte class changes the gas-phase logic. Cross-domain projects often need different analytical support for exactly that reason, whether the bottleneck is protein-level identification in complex discovery datasets or targeted lipid readouts for class-resolved structural interpretation.
Fragmentation methods differ because energy enters the ion differently
CID, HCD, ETD, ECD, and UVPD are often presented as a menu of tandem MS options. That presentation hides the real issue. These methods are not just different names for "fragmenting harder" or "fragmenting softer." They differ because they deposit energy through different physical routes, on different time scales, and with different consequences for charge redistribution and bond-selective cleavage.
Once that is clear, their spectra stop looking arbitrary.
CID: slow vibrational heating favors familiar backbone channels
Collision-induced dissociation remains the clearest example of how gradual vibrational heating can steer a precursor toward a relatively narrow family of dissociation pathways. In CID, the ion undergoes repeated low-energy collisions with neutral gas molecules. Each collision contributes a small amount of internal energy. That energy accumulates as vibrational excitation until one or more dissociation routes become competitive.
In peptide analysis, this often favors b- and y-type ions because proton mobility can activate the amide backbone and lower the barrier for those cleavage channels. CID therefore does not produce random damage. It produces selective dissociation under slow heating.
Wells and McLuckey's overview remains useful here because it emphasizes CID as a mechanistic process rather than a simple instrument setting. The ion climbs an energy landscape. The pathways that open first, or dominate most efficiently, determine what is observed.
Why CID often collapses into neutral loss before it yields full backbone coverage
CID is powerful, but it is selective. If the lowest-barrier route for the activated precursor is a small neutral elimination rather than productive backbone cleavage, that channel can dominate. This is why some modified peptides, especially labile PTM-bearing species, spend much of their signal budget on neutral loss instead of sequence-informative ions.
That behavior should not be overclaimed either way. It does not mean CID is wrong. It means CID is faithfully reporting the lowest accessible path under that activation history. For PTM-rich samples, the practical question is therefore not whether CID works in principle, but whether CID preserves the information you most need.
HCD: beam-type collision changes both fragment balance and interpretive utility
HCD is often described as a stronger version of CID. That is directionally correct but chemically too shallow. HCD is better understood as beam-type collisional activation, where ions are accelerated into a collision region and undergo a different effective energy-delivery history. The output often still includes strong b/y content for peptides, but the balance shifts.
Low-mass ions become more visible. Reporter ions become practical. Internal fragments may appear more readily. Secondary fragmentation can become more pronounced depending on the setting and precursor class. So HCD is not simply "more fragmentation." It is a different tradeoff between backbone coverage, low-mass visibility, and secondary dissociation.
That is exactly why HCD became so useful in quantitative proteomics. Reporter-ion generation depends on that low-mass access. At the same time, HCD is not automatically superior for every analytical question. If the priority is preserving fragile chemistry, stronger collision-driven activation may still route too much signal into loss pathways before the desired evidence is preserved.
So the better question is not whether HCD is better than CID. The better question is what information each mode tends to preserve or destroy under the actual precursor chemistry being studied.
In applied work, that difference often shapes whether a workflow is optimized around isobaric-label quantification and reporter-ion recovery or around PTM-focused downstream interpretation support.
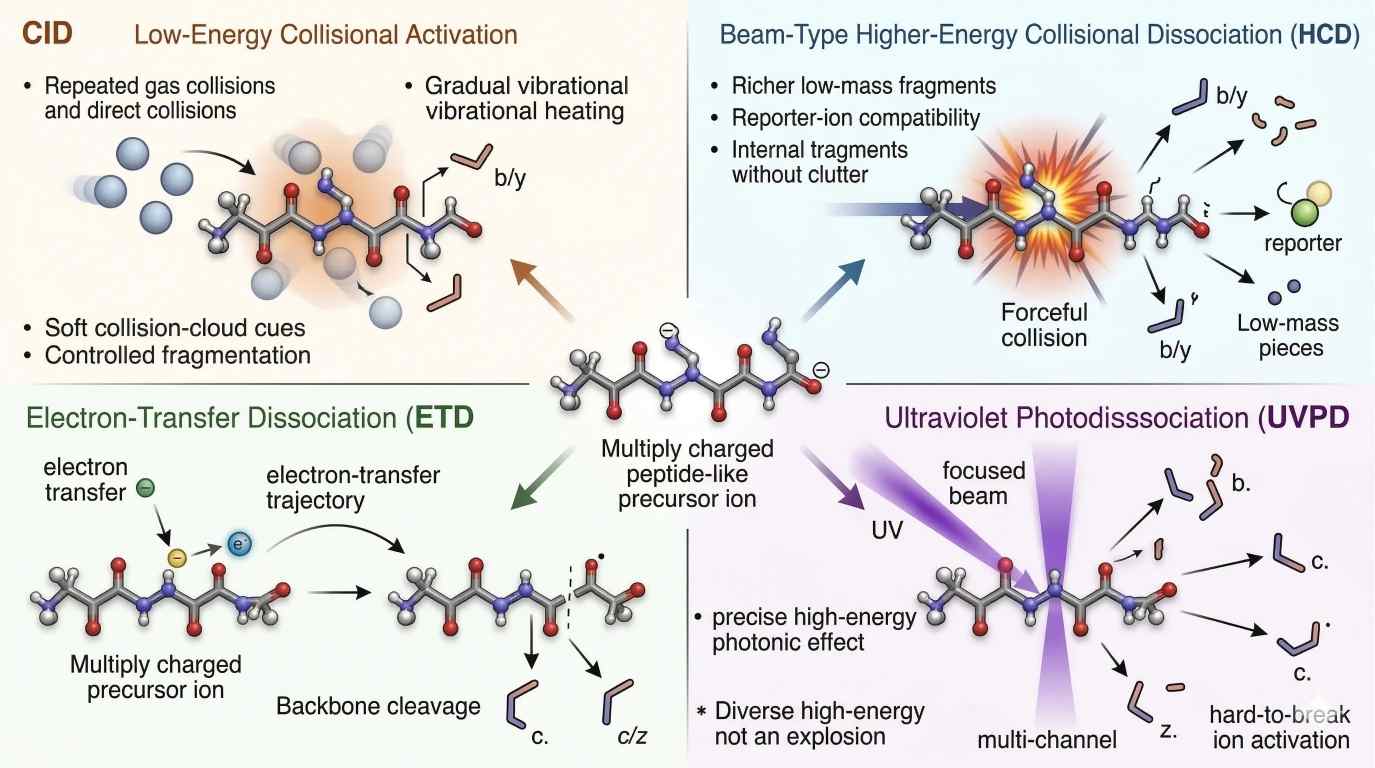 Figure 2. Comparative fragmentation physics of CID, HCD, ETD/ECD, and UVPD. Design this as a side-by-side mechanism map. For each mode, show the energy input type, the characteristic time scale, the dominant fragment bias, and the tendency to retain or lose labile PTMs. Make CID and HCD visually distinct by showing slow heating versus beam-type collision. Make ETD/ECD electron-driven. Make UVPD photon-driven. The takeaway should be immediate: the same precursor does not pass through one universal fragmentation regime.
Figure 2. Comparative fragmentation physics of CID, HCD, ETD/ECD, and UVPD. Design this as a side-by-side mechanism map. For each mode, show the energy input type, the characteristic time scale, the dominant fragment bias, and the tendency to retain or lose labile PTMs. Make CID and HCD visually distinct by showing slow heating versus beam-type collision. Make ETD/ECD electron-driven. Make UVPD photon-driven. The takeaway should be immediate: the same precursor does not pass through one universal fragmentation regime.
ETD and ECD move the precursor into radical chemistry
Electron-transfer dissociation and electron-capture dissociation matter because they change the precursor's electron character. Instead of relying on slow collisional heating, they generate radical intermediates. That shift opens cleavage routes that differ from the closed-shell pathways favored in CID and HCD.
In peptides and proteins, this commonly leads to c- and z-type ions. More importantly, it often allows backbone cleavage to proceed while labile modifications remain attached to the fragments. That is why ETD and ECD became so important for modification-rich systems. They do not guarantee perfect spectra, but they often preserve the kind of evidence that collisional activation tends to lose.
The original ETD paper by Syka and colleagues made this especially clear by showing that electron transfer to multiply protonated peptides induced backbone fragmentation analogous to ECD-like behavior, but in a format practical for ion trap workflows. (PNAS)
Why electron-based methods are strongest for highly charged precursors
ETD and ECD are not equally effective for every ion. They tend to perform best when the precursor carries enough charge to support efficient electron transfer or capture and to retain informative fragment ions after cleavage. That charge dependence is one reason they are especially useful for highly charged peptides and intact proteins.
This is also why electron-based methods are so often linked to top-down strategies and PTM-focused analysis. Large ions carry multiple charges and often contain the exact fragile chemistry that collisional modes struggle to preserve. In such cases, radical-driven cleavage is not just an alternative. It can be the only route that keeps backbone evidence and modification evidence in the same spectrum.
That logic underlies many glycopeptide characterization workflows and top-down PTM characterization strategies, where the core problem is not just fragmentation efficiency, but what kind of information survives fragmentation at all.
UVPD is photon-driven, not a harsher version of HCD
Ultraviolet photodissociation belongs in its own category because photons, not collisions or electron transfer, provide the activation energy. Brodbelt's review remains a key reference here because it frames photodissociation as a distinct activation strategy that can provide broad and versatile fragmentation for biological molecules.
That distinction matters. UVPD is not just a more forceful HCD method. A 193 nm photon can open high-energy channels that may be weak or inaccessible under slow collisional heating. The resulting fragment ensemble can be broader, sometimes denser, and often especially useful for analytes that resist clean fragmentation by other means.
For intact proteins and structurally stubborn precursors, that can translate into richer sequence coverage. But the key interpretive point is simpler: UVPD changes the activation logic itself. Once the energy source changes from collision to photon, the spectrum should not be judged as if it came from the same pathway family as CID or HCD.
Neutral loss is one of the most useful and most misused concepts in tandem MS. It is useful because some losses recur often enough to guide interpretation. It is misused because analysts often treat those losses as if they were direct structural proof. They are not. A neutral loss tells you what the activated precursor found easiest to shed under a given energy regime. That can be highly informative, but it is still only one layer of evidence.
Water loss, 18.01 Da, often appears when the precursor contains accessible hydroxyl-bearing or dehydration-prone functionality. Ammonia loss, 17.03 Da, commonly reflects routes available to amine-bearing structures or specific side-chain environments. Phosphoric acid loss, 97.97 Da, is especially important in phosphopeptide work because phosphoserine- and phosphothreonine-containing precursors often show strong loss pathways under collisional activation. The temptation is to treat these losses as answers. The disciplined approach is to treat them as ranked clues. A phosphate-related neutral loss may strongly support precursor lability, but it does not by itself localize the site. A dehydration peak may support functional-group plausibility, but it does not prove which hydroxyl-bearing region was involved.
That distinction becomes even more important when collision energy changes. A neutral-loss-dominated spectrum does not always mean the precursor is poorly chosen or the acquisition failed. It may simply mean that the lowest-energy path consumed the signal before more structure-rich channels became competitive. This is why neutral loss should be read as a report on activated stability, not as a substitute for backbone evidence, site evidence, or orthogonal confirmation.
Diagnostic ions work the same way. They can be extremely powerful, but their evidential rank depends on what exactly they define. Some are class-defining. Some are substructure-defining. Very few are full-structure-defining on their own.
In lipidomics, the phosphocholine fragment at m/z 184 is the classic example. It can strongly support a choline-containing lipid class, yet it does not on its own resolve sn-position, full acyl arrangement, or double-bond placement. In metabolomics, certain low-mass ions can strongly support a recurring substructure, but the rest of the skeleton may still remain unresolved. The best interpreters therefore do not ask only whether a diagnostic ion is present. They ask what level of structural claim that ion can honestly support.
That is why confident annotation usually depends on evidence layering. Exact mass narrows the possibilities. Isotopic fit constrains the formula space. Adduct behavior adds precursor chemistry. Diagnostic ions add class or substructure support. Broader fragmentation logic tests whether the rest of the spectrum belongs to the same explanation. In discovery settings, this is often where LC-MS/MS untargeted metabolite profiling and unknown-metabolite identification workflows begin to diverge in value, because one is optimized for broad detection while the other is optimized for narrowing structure from layered evidence.
Collision energy is often treated as a simple intensity knob. In practice, it is a pathway-selection variable. Changing collision energy does not merely scale one spectrum up or down. It changes which dissociation routes become competitive, how much precursor survives, whether low-mass ions appear, and when secondary fragmentation begins to reshape the product-ion family.
That is why the same precursor can produce markedly different spectra across collision settings even when the m/z is unchanged. At low energy, one or two facile losses may dominate. At moderate energy, a more useful fragment ladder may emerge. At higher energy, internal fragments, reporter ions, or secondary products may become prominent. Past that point, informative peaks may begin to erode. A single collision-energy setting should therefore not be overclaimed as the one "true" spectrum of an analyte. It is only one view of the analyte's fragmentation landscape.
This is also why collision-energy ramps can be so useful. A ramp samples a broader window of pathway competition and often recovers both lower-threshold and higher-threshold fragmentation events in one acquisition strategy. The benefit is broader evidence. The cost is that the resulting spectrum can become more composite, especially when facile neutral losses and productive backbone cleavage coexist.
Mirror plots are valuable for exactly the same reason. They make similarity visible, but they can also exaggerate confidence. A strong mirror match may be driven by a small set of dominant peaks. A weaker match may arise not from misidentification, but from different activation histories. Poor mirror agreement should not be overinterpreted as a wrong identification when the precursor has been activated under clearly different energy-delivery conditions. The comparison is only as fair as the activation history behind it.
This matters most across platforms. Even when two systems use the same method label, they may not deliver the same internal energy history. Isolation width, gas environment, timing, and collision-energy calibration all shape the observed fragment balance. In practice, this is one reason why integrated bioinformatics support for proteomics interpretation and data preprocessing and normalization workflows for complex omics datasets become so important once spectra are compared across runs, batches, or instruments rather than interpreted in isolation.
Chimeric spectra make these problems harder, not because they are rare, but because they are common enough to hide inside apparently convincing matches. Isolation windows have finite width. Co-eluting species overlap. Similar m/z values are captured together. The resulting MS/MS spectrum may therefore contain fragments from more than one precursor.
The real danger is not that a chimeric spectrum looks obviously messy. The danger is that it can look partly coherent. One subset of peaks may support a plausible match, while another subset comes from a different precursor and quietly degrades localization confidence, structural specificity, or sequence interpretation. Analysts who rely only on one search score can miss that split.
A better inspection sequence is straightforward. First, ask whether the main ion series behaves like one coherent family. Second, check whether charge-state logic remains internally consistent. Third, look for unexplained dominant peaks that do not fit the proposed precursor chemistry. If those checks fail, co-isolation should be treated as a live explanation rather than an afterthought.
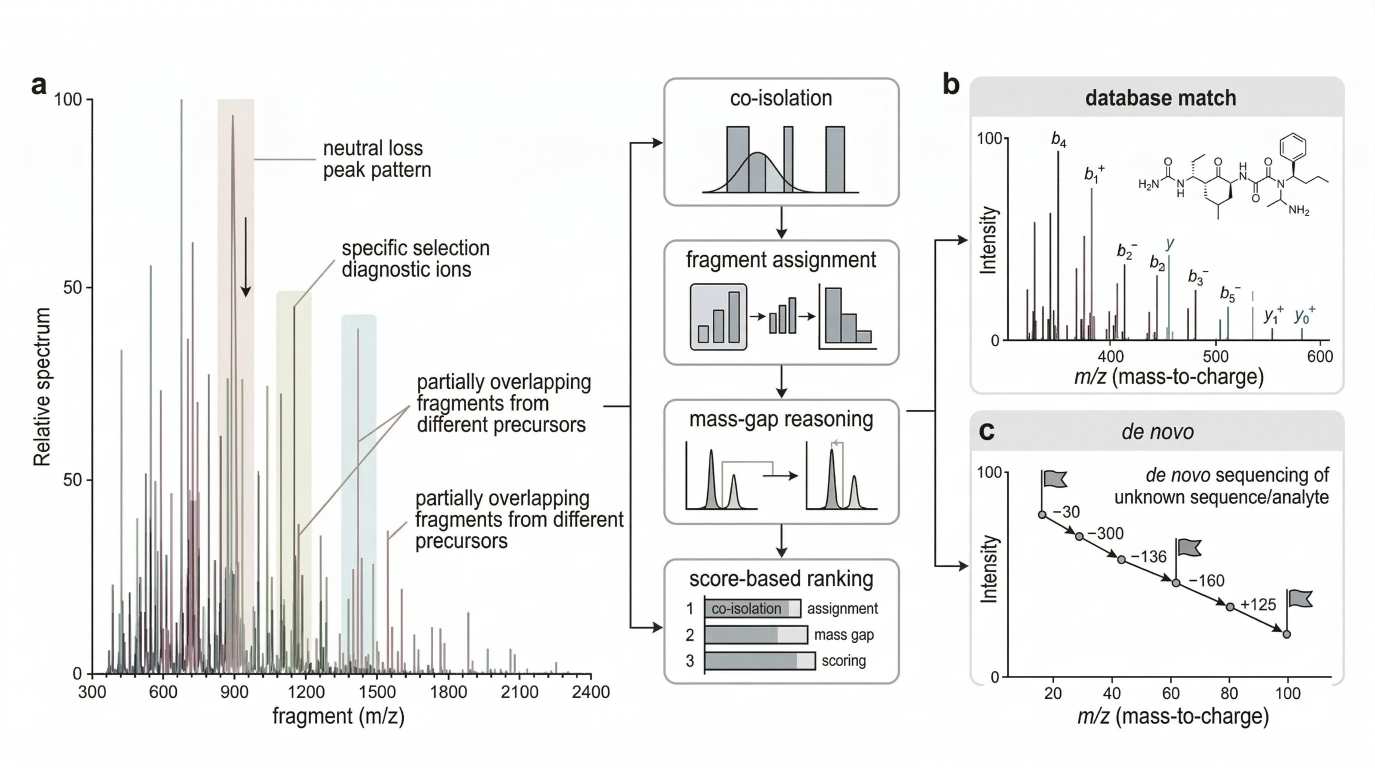 Figure 3. Neutral loss, evidential rank, and chimeric-spectrum deconvolution. Build this figure in three panels. Left: show neutral loss as a clue rather than proof, with one precursor yielding strong small-molecule losses but incomplete structural resolution. Center: show a compact evidential-rank pyramid, separating class markers, substructure markers, backbone-defining ions, and full-structure evidence. Right: show co-isolation leading to a mixed spectrum, followed by deconvolution checks based on ion-series coherence, charge consistency, and unexplained dominant peaks. The takeaway should be that a spectrum is an evidence hierarchy, not a flat peak list.
Figure 3. Neutral loss, evidential rank, and chimeric-spectrum deconvolution. Build this figure in three panels. Left: show neutral loss as a clue rather than proof, with one precursor yielding strong small-molecule losses but incomplete structural resolution. Center: show a compact evidential-rank pyramid, separating class markers, substructure markers, backbone-defining ions, and full-structure evidence. Right: show co-isolation leading to a mixed spectrum, followed by deconvolution checks based on ion-series coherence, charge consistency, and unexplained dominant peaks. The takeaway should be that a spectrum is an evidence hierarchy, not a flat peak list.
Search engines are powerful because they simplify chemistry into scoreable evidence. That is their strength and also their main limitation.
Mascot-style logic asks, in broad terms, how likely the observed agreement between a candidate and a spectrum could arise by chance. SEQUEST-style logic asks how well the observed and theoretical fragment patterns correlate. Both approaches are extremely effective when the spectrum behaves close to the model: one dominant precursor, coherent ion series, reasonable noise, and fragmentation that largely follows expected routes. The foundational papers by Eng et al. and Perkins et al. remain important because they formalized these two major scoring traditions. (ScienceDirect)
But score is not chemistry. A high score does not prove that every observed peak belongs to one precursor. It proves that the overall match to the scoring model is strong. That distinction matters in mixed spectra, in neutral-loss-dominated spectra, and in spectra that contain strong internal fragments or activation-mode-specific behavior that the scoring model only approximates.
A useful manual review step is therefore simple. Ask whether the score is supported by ion-series coherence, charge-state consistency, and the absence of strong unexplained peaks. When those three checks fail, the spectrum may still be score-rich but chemically incomplete.
De novo sequencing asks a harder question. Database search begins with a candidate list. De novo interpretation does not. It must infer sequence or structure from fragment evidence itself. In peptide work, the classic mass-gap idea is central: if adjacent fragment differences map cleanly onto residue masses, sequence information can be assembled even without a database match.
That strategy is powerful because it can recover structure from incomplete ladders, novel peptides, and unexpected sequence changes. It is also fragile because missing ions, internal fragments, and chimeric interference can distort the gap pattern. For that reason, de novo interpretation is not just a fallback for failed database search. It is a different evidence framework.
The same logic extends conceptually into unknown small-molecule work, although the search space is much larger. In both cases, the analyst is no longer asking which known candidate best fits the whole spectrum. The analyst is asking which local mass relationships can be assembled into a chemically coherent explanation.
That is where de novo antibody-sequencing workflows and top-down sequencing strategies for intact or large proteoforms solve a different problem from routine candidate matching. They are built for spectra that must be interpreted as structural evidence first and database hits second.
The most reliable tandem MS interpretation therefore does not force every peak to carry the same meaning. It separates evidence by role. Exact mass, isotopic pattern, adduct state, and charge state define the precursor layer. Activation mode defines the physical regime. Fragment ions then separate into evidential ranks: class markers, substructure markers, backbone-defining ions, neutral losses, and byproducts. Search scores add one more layer, but they do not erase the need for chemical review.
Once that hierarchy is clear, method choice also becomes clearer. The question is not which fragmentation mode is best in the abstract. The question is which mode preserves the information you most need.
If the goal is routine peptide identification with familiar, score-friendly ladders, CID or HCD may be ideal. If the goal is isobaric quantification, HCD becomes especially attractive because reporter ions matter. If the goal is preserving labile PTMs on highly charged peptides, ETD or related hybrid methods often become more informative because backbone cleavage can occur without consuming the evidence in slow-loss channels. If the target is a large or fragmentation-resistant proteoform, UVPD may open channels that collisional activation never accesses efficiently. Electron-transfer dissociation, electron-capture dissociation, and UV photodissociation are now all established complements to collision-driven methods rather than niche curiosities.
Here is the practical comparison in compact form:
| Parameter | CID | HCD | ETD |
|---|---|---|---|
| Dominant activation logic | Low-energy vibrational heating | Beam-type collisional activation | Electron-transfer radical activation |
| Common peptide fragment bias | b/y ions | b/y ions with stronger low-mass visibility | c/z ions |
| PTM retention | Often limited for labile PTMs | Better in some contexts, but still collision-driven | Strong for many labile PTMs |
| High-mass / highly charged precursor utility | Moderate | Moderate to strong | Strong when charge is high |
| Reporter-ion compatibility | Limited | Excellent | Limited |
| Routine duty-cycle practicality | Widely used | Widely used and versatile | More charge-dependent |
The table is useful, but it only works when its physics stays in view. CID and HCD are not defined only by their favored fragment names. They are defined by how energy enters the ion. ETD is not defined only by c/z output. It is defined by radical initiation. Once that is clear, tandem MS stops looking like a menu of acronyms and starts looking like a controlled set of gas-phase experiments.
The broad conclusion is simple. Fragmentation is not random damage. It is structured gas-phase behavior. A precursor forms in a specific ionic state. Charge is held in a specific topology. Energy is delivered through a specific mechanism. Only then do dissociation routes compete. The spectrum is the record of that sequence.
FAQ
What does a strong neutral-loss peak actually prove?
Usually less than people think. It proves that a small-molecule elimination pathway was energetically accessible and competitive under the chosen activation regime. It may support a functional-group class or PTM lability, but it does not by itself prove site localization or full structure.
Why can the same precursor produce different spectra across runs?
Because the precursor may not have experienced the same activation history. Small changes in collision energy, isolation width, gas conditions, or co-isolation burden can redistribute which pathways dominate, even when the nominal precursor m/z is identical.
When should a poor mirror match not be overinterpreted?
When the spectra were acquired under clearly different activation histories or on different platforms with non-equivalent collision-energy behavior. In that case, lower similarity may reflect pathway weighting rather than wrong identity.
Why do some peptides give complete b/y ladders and others do not?
Because backbone fragmentation depends on charge topology, proton mobility, local sequence environment, and pathway competition. A peptide with trapped protonation or dominant neutral-loss channels may remain only partially ladder-like even when the instrument is working correctly.
Why is ETD often preferred for modification-rich peptides?
Because ETD generates radical intermediates and often allows backbone cleavage while retaining labile modifications on the fragment ions. That makes it especially useful when PTM site analysis is more important than reporter-ion intensity or routine collisional fragmentation.
Is a diagnostic ion enough for a confident annotation?
Often enough for class assignment, rarely enough for full structural resolution. Diagnostic ions should be ranked by what they truly define: class, substructure, or complete structure.
What is the main danger of a chimeric spectrum?
Not that it is obviously messy, but that it can look partly correct. One precursor may explain enough peaks to score well while a second precursor quietly contaminates localization or structural interpretation.
When is de novo sequencing more useful than database searching?
When the analyte may not exist in the reference database, when sequence novelty is expected, or when the fragment ladder contains enough local mass-gap information to support structural inference even without a whole-sequence candidate.
References
- Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24(4):508-548. DOI: 10.1002/mas.20024
- Wells JM, McLuckey SA. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 2005;402:148-185. DOI: 10.1016/S0076-6879(05)02005-7
- Wysocki VH, Resing KA, Zhang Q, Cheng G. Mass spectrometry of peptides and proteins. Methods. 2005;35(3):211-222. DOI: 10.1016/j.ymeth.2004.08.013
- Reid GE, McLuckey SA. 'Top down' protein characterization via tandem mass spectrometry. J Mass Spectrom. 2002;37(7):663-675. DOI: 10.1002/jms.346
- Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101(26):9528-9533. DOI: 10.1073/pnas.0402700101
- Zubarev RA. Electron-capture dissociation tandem mass spectrometry. Curr Opin Biotechnol. 2004;15(1):12-16. DOI: 10.1016/j.copbio.2003.11.002
- Brodbelt JS. Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem Soc Rev. 2014;43(8):2757-2783. DOI: 10.1039/C3CS60444F
- Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:105. DOI: 10.1186/1471-2105-8-105
- Eng JK, McCormack AL, Yates JR III. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976-989. DOI: 10.1016/1044-0305(94)80016-2
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551-3567. DOI: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2
- Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5(4):397-409. DOI: 10.1007/BF01049915
- Hsu FF, Turk J. Structural determination of glycosphingolipids as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisional-activated dissociation on a triple stage quadrupole instrument. J Am Soc Mass Spectrom. 2001;12(1):61-79. DOI: 10.1016/S1044-0305(00)00194-X
This content is intended for research-use analytical interpretation only and does not support clinical diagnosis or therapeutic decision-making.














