Affinity Purification Mass Spectrometry (AP-MS) Service
Affinity Purification Mass Spectrometry (AP-MS) is a gold-standard approach for mapping protein-protein interactions (PPIs) with high sensitivity, specificity, and physiological relevance.
At Creative Proteomics, our AP-MS service is designed to support researchers seeking reliable, high-resolution insights into protein complex composition, dynamics, and function.
Whether you're investigating stable or transient interactomes, our customizable workflow enables precise protein purification mass spectrometry analysis tailored to your experimental system. With robust affinity enrichment strategies and high-resolution MS platforms, we deliver reproducible data that drives confident biological interpretation.
Our service is ideal for pharmaceutical teams, academic labs, and CROs focused on interactome characterization, drug target validation, or systems biology research.
Submit Your Request Now
×
- Comprehensive MS Report
- High-Confidence Interaction Lists
- Functional Enrichment Analysis
- Protein Interaction Network Maps
- Why Choose AP-MS
- Service Overview
- Advantage
- Application
- Delivery&Demo
- Sample Requirement
- FAQ
- Case Study
- Publication
Why Choose AP-MS for Protein-Protein Interaction Studies?
Understanding PPIs is essential for unraveling cellular mechanisms, signaling pathways, and complex regulatory networks. AP-MS stands out as a powerful technique for interactome research due to its ability to capture both stable and transient protein complexes under near-physiological conditions.
Compared to traditional interaction mapping methods like yeast two-hybrid or pull-down assays, AP-MS—especially when integrated with Co-Immunoprecipitation (Co-IP)—offers superior specificity, scalability, and biological relevance. This approach minimizes background noise and allows for a more accurate reflection of native cellular environments.
Key advantages of AP-MS include:
- Detection of both stable and transient interactions, enabling a comprehensive view of dynamic protein assemblies.
- Low background and high signal-to-noise ratio, thanks to optimized purification and MS protocols.
- Compatibility with high-throughput workflows, supporting large-scale interactome profiling.
- Flexible experimental design, including use of various affinity tags (e.g., FLAG, HA, His) or tag-free strategies.
Our AP-MS Capabilities
Sample Type Compatibility
We accept a wide range of input materials, including cultured cell lines, primary cells, tissues, and purified recombinant proteins. This allows researchers to study PPIs under native or overexpression conditions based on project needs.
Affiliation Tag Versatility
Our protocols support a broad spectrum of affinity tags such as FLAG, HA, His, Myc, and biotin, among others. We can also accommodate tag-free approaches when working with high-quality antibodies, enabling flexibility for various protein systems.
High-Resolution Mass Spectrometry Platforms
We leverage state-of-the-art LC-MS/MS systems, including high-resolution Orbitrap instruments, to ensure deep proteome coverage and accurate peptide identification.
Quantitative Strategies
Both label-free quantitation and isobaric labeling approaches (e.g., TMT, iTRAQ) are available to support comparative interactome analysis and condition-specific studies.
Advanced Bioinformatics Support
Our integrated bioinformatics pipeline includes:
- Interaction confidence scoring and filtering
- Statistical analysis for differential interactors
- Functional enrichment analysis (GO, KEGG)
- Network visualization and annotation
For studies requiring enhanced purification specificity, our Tandem Affinity Purification (TAP-MS) service offers a sequential dual-tag system that minimizes background while maintaining high recovery of interaction partners.
Workflow Overview: From Sample to Insight
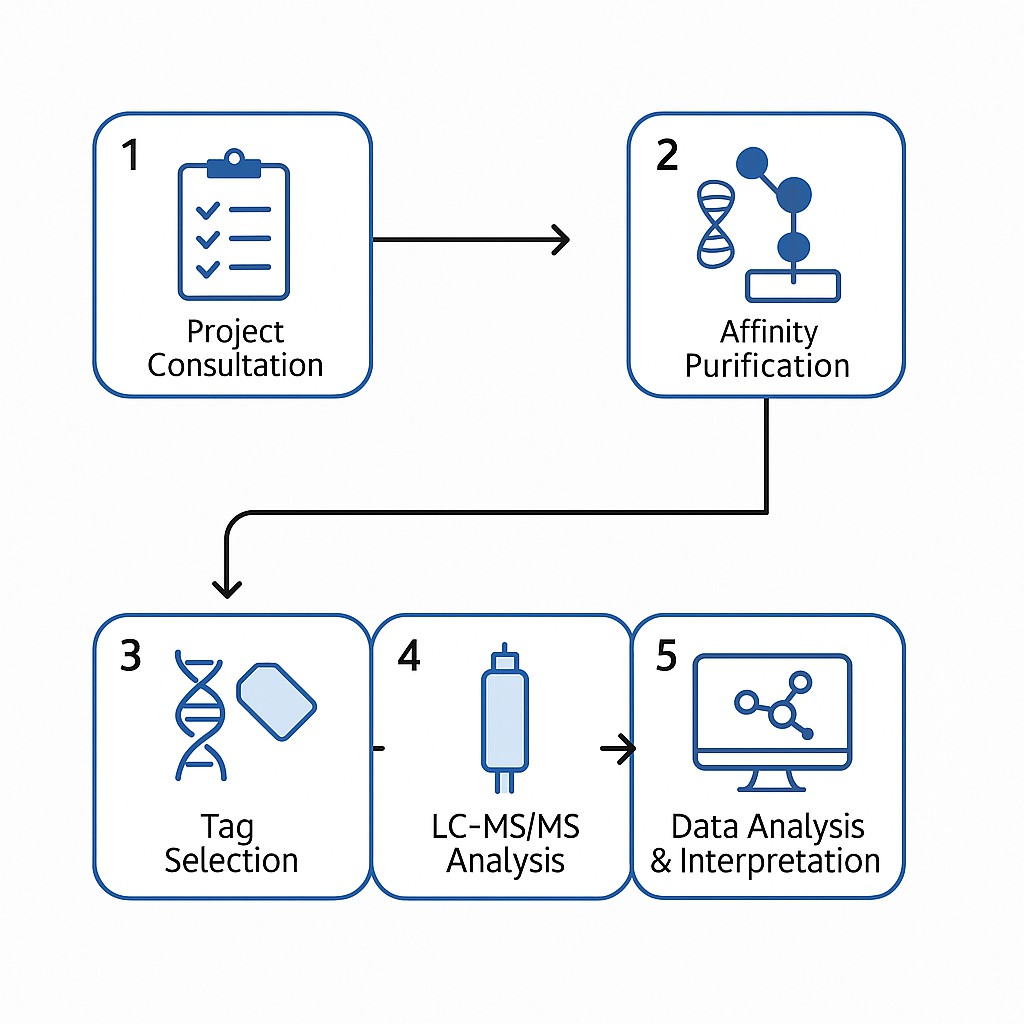
1. Project Consultation and Experimental Design
Every study begins with a detailed consultation to understand your biological questions, sample availability, and preferred affinity strategy. We assist in selecting optimal controls, expression systems, and experimental conditions tailored to your goals.
2. Tag Selection and Expression System Optimization
Based on target protein characteristics and interaction dynamics, we help you choose the most appropriate tag (e.g., FLAG, His, HA) and expression platform (e.g., mammalian cells, yeast, bacteria). For native protein studies, we also offer antibody-based Co-IP support.
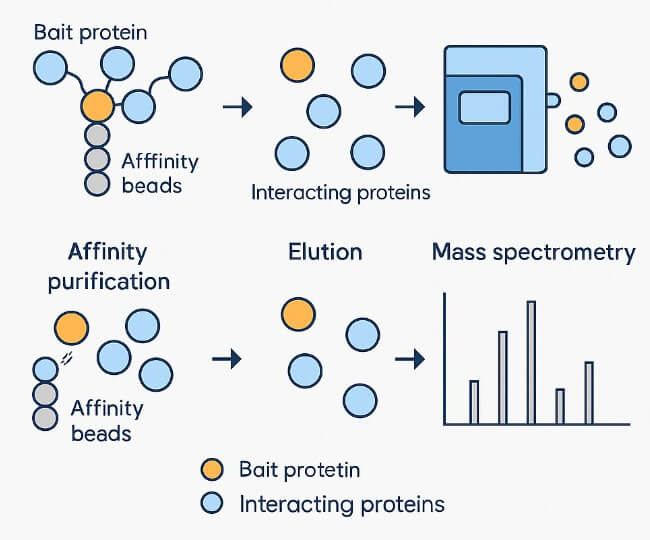
3. Affinity Purification of Protein Complexes
Using optimized protocols for low-background enrichment, we isolate protein complexes under conditions that preserve native interactions—capturing both strong and weak interactors with high specificity.
4. LC-MS/MS Analysis
Purified samples are analyzed on high-resolution Orbitrap platforms using data-dependent acquisition (DDA) or data-independent acquisition (DIA) modes, depending on project needs. Our QC benchmarks ensure consistent peptide coverage and mass accuracy.
5. Bioinformatics and Network Construction
Raw MS data are processed through a robust pipeline that includes protein identification, background filtering, quantification (if applicable), and protein-protein interaction network modeling.
6. Data Delivery and Expert Interpretation
Clients receive a comprehensive data package including processed spectra, interaction tables, enrichment results, and network maps. A follow-up consultation is offered to guide data interpretation and integration into your broader research.
For expanded interactome studies, downstream analysis can be further enhanced with our IP-MS protein interactomics solution, which integrates large-scale interaction profiling with biological context filtering and visualization tools.
Why Choose Creative Proteomics?
Proven Expertise: A dedicated team with deep experience in protein interactomics ensures your project is designed and executed flawlessly.
Cutting-Edge Technology: We use the latest Orbitrap MS and optimized purification methods to achieve unmatched sensitivity and specificity.
Tailored Solutions: Custom workflows fit your exact needs—from rare samples to high-throughput studies—maximizing research impact.
Uncompromising Quality: Rigid quality control protocols guarantee reliable, reproducible data you can confidently publish.
One-Stop Service: From AP-MS to Co-IP and TAP-MS, our integrated platform simplifies complex protein interaction analyses.
Global Trust: Leading academic labs and pharma R&D worldwide choose us for accurate, actionable PPI data.
Application Areas
Drug Target Identification and Validation
AP-MS enables precise mapping of target protein interactors, helping pharmaceutical researchers uncover binding partners and pathways critical for drug efficacy and mechanism of action studies.
Signal Transduction Pathway Analysis
Dissect complex signaling cascades by identifying dynamic protein complexes that regulate cellular responses, facilitating deeper understanding of pathway modulation under physiological and experimental conditions.
Epigenetic Regulation Mechanism Studies
Characterize protein assemblies involved in chromatin remodeling, histone modification, and transcriptional regulation to unravel epigenetic control networks.
Cell Cycle and Stress Response Research
Explore interaction networks governing cell cycle checkpoints, DNA repair, and stress adaptation to shed light on cellular homeostasis and resilience mechanisms.
Biomarker Discovery
Identify novel protein partners associated with disease-related pathways or cellular states, providing candidates for biomarker development and translational research.
Deliverables
High-Quality Raw and Processed Mass Spectrometry Data
Including raw spectra files and processed peptide/protein identification reports, ensuring full transparency and data reproducibility.
Detailed Protein Identification and Interaction Lists
Comprehensive tables listing all detected proteins with quantitative values (when applicable), filtered to highlight confident interaction partners.
Functional Enrichment Analysis Reports
Gene Ontology (GO) and KEGG pathway enrichment analyses to contextualize interactors within biological processes and pathways relevant to your study.
Protein-Protein Interaction Network Visualizations
Intuitive network graphs and annotation files that facilitate the exploration of interaction landscapes and hypothesis generation.
Technical Interpretation Support
Optional one-on-one consultation sessions with our proteomics experts to discuss data insights, troubleshoot issues, and assist with integration into your ongoing research.
Demo

Sample Requirement
| Sample Type | Recommended Sample Amount |
|---|---|
| Cultured cell pellets (adherent or suspension cells) | Typically 10^7 to 10^8 cells per replicate, depending on protein abundance |
| Tissue samples (fresh or flash-frozen) | Generally 50–200 mg per replicate |
| Recombinant proteins or protein complexes | Amount varies; please consult for project-specific advice |
| Primary cells (subject to consultation) | Amount varies; consult prior to submission |
Frequently Asked Questions (FAQs)
What types of samples can be used for AP-MS?
Our service supports diverse sample types including cultured cell lines, primary cells, tissue extracts, and recombinant proteins, providing flexibility for various research models.
Is it necessary to have a tagged protein for affinity purification?
While affinity tags like FLAG, His, or HA improve purification efficiency, we also offer antibody-based Co-Immunoprecipitation (Co-IP) options for native protein studies without tagging.
Can you perform quantitative analysis in AP-MS experiments?
Yes, we support multiple quantification methods such as label-free quantitation and isobaric tagging (TMT/iTRAQ) to provide robust comparative data.
What is the minimum amount of protein required for analysis?
Sample input requirements depend on protein abundance and purification strategy. We will provide personalized recommendations during project consultation to ensure optimal results.
Can I request purification only without mass spectrometry analysis?
Our AP-MS service is designed as an integrated workflow combining purification and mass spectrometry for best data quality. Please contact us if you have specific purification-only needs.
How is data confidentiality maintained?
We adhere to strict data privacy and confidentiality protocols, ensuring your research data is securely handled and not shared without permission.
Are replicate experiments supported?
Yes, replicate analyses are strongly recommended for reliable interaction validation and are fully supported by our quantitative workflows.
What kind of data will I receive?
You will get raw and processed mass spectrometry files, protein identification and quantification tables, enrichment analysis reports, and network visualization files, along with expert interpretation support if requested.
Learn about other Q&A.
Case Study: High-Throughput Protein–Protein Interaction Mapping Using Orbitrap–Astral MS

J Proteome Res. 2025 Mar 3;24(4):2006–2016. doi: 10.1021/acs.jproteome.4c01040
- Background
- Methodology
- Results
- Conclusion
Traditional Affinity Purification Mass Spectrometry (AP-MS) methods are limited by throughput constraints, typically requiring 20–90 minutes per sample. This limitation hinders large-scale protein–protein interaction (PPI) studies.
The study utilized the Thermo Scientific Orbitrap–Astral mass spectrometer, which integrates an Asymmetric Track Lossless (Astral) analyzer for enhanced ion detection. Coupled with a rapid 7-minute high-flow liquid chromatography (LC) separation, the system enabled the analysis of 216 AP-MS samples in approximately 29 hours.
The approach identified 998 confident PPIs between 59 viral and 998 human proteins, achieving a median of eight interactions per bait. This high-throughput method demonstrated reproducibility and sensitivity, with a median coefficient of variation of 21.6% for protein quantification across replicates.
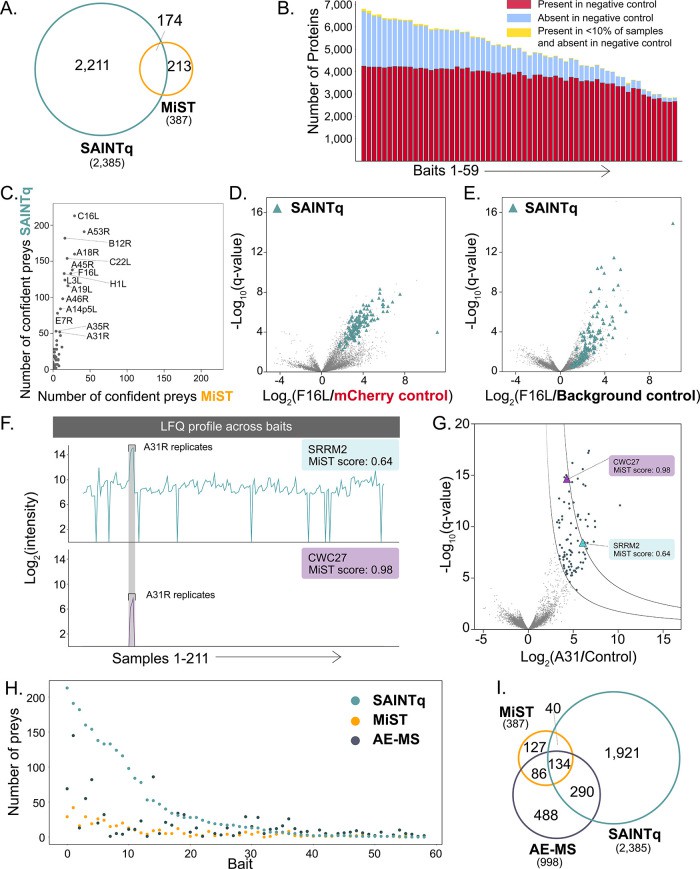 Characterization and investigation of scoring software discrepancies
Characterization and investigation of scoring software discrepancies
The combination of rapid LC separation and the Orbitrap–Astral MS system significantly accelerates PPI mapping, facilitating large-scale interactome studies. This advancement holds promise for more comprehensive biological insights and drug discovery applications.
Publication
Here are some publications in PPIs research from our clients:

- Protein interactors of Spindle Pole Body (SPB) components and septal proteins in fungus Neurospora crassa: A mass spectrometry-based dataset. Data in Brief. 2024. https://doi.org/10.1016/j.dib.2023.109980
- Morphological and genetic screens reveal mechanisms of BiDAC-induced plasma membrane protein degradation.Research Square. 2024. https://doi.org/10.21203/rs.3.rs-4438596/v1
- Characterizing the proteome of bullous pemphigoid blister fluid utilizing tandem mass tag labeling coupled with LC–MS/MS.Archives of Dermatological Research. 2022. https://doi.org/10.1007/s12016-017-8633-4











