Elucidating protein phosphorylation's complex dynamics is essential for deciphering cellular signaling networks, pathological processes, and druggable targets. Phosphoproteomics provides the critical blueprint for this exploration, yet translating biological specimens into biologically meaningful datasets demands adherence to a rigorously optimized analytical pipeline. This technical guide details each procedural phase, empowering laboratory specialists to refine methodological execution while ensuring contract research organization (CRO) clients comprehend workflow stages and anticipated deliverables.
Primary Objective: Enable comprehensive profiling of phosphorylated peptides—including precise site localization—within intricate biological matrices.
Central Technical Challenge: Phosphomodifications exhibit transient dynamics, typically occur at low stoichiometry, and undergo signal suppression by dominant non-phosphorylated peptide populations. Consequently, enrichment methodologies demonstrating exceptional sensitivity and specificity constitute an imperative methodological requirement.
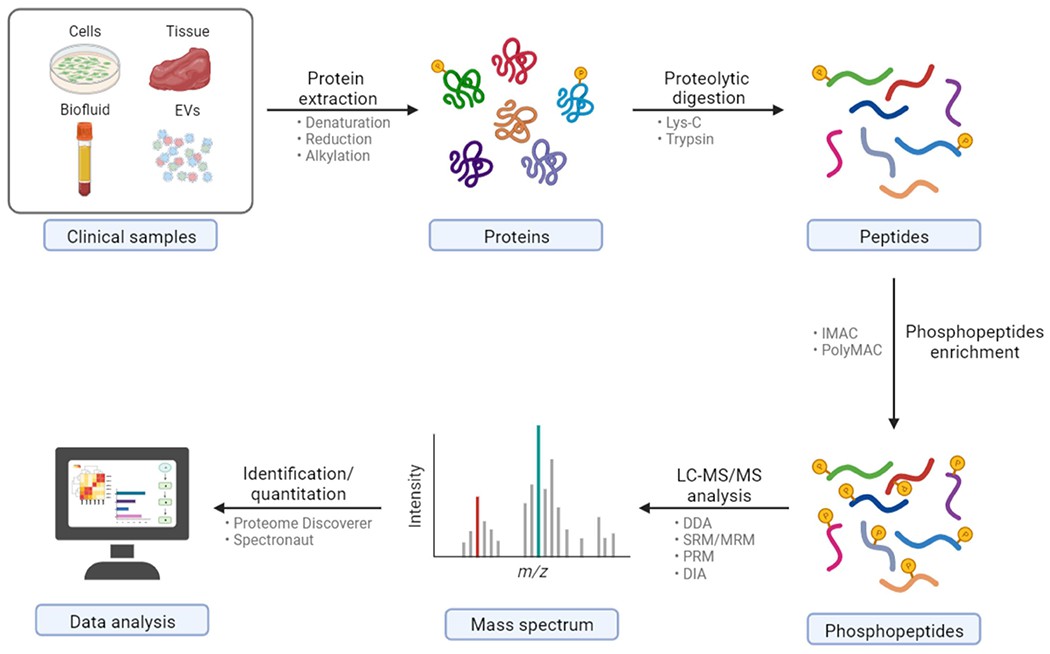 General workflow of clinical sample preparation for bottom-up phosphoproteomics via LC-MS/MS (Wu X et al., 2023)
General workflow of clinical sample preparation for bottom-up phosphoproteomics via LC-MS/MS (Wu X et al., 2023)
Workflow: Detailed explanation step by step
Sample Preparation: Foundational Integrity Preservation
Core Principle: Maintain phosphoprotein integrity through immediate enzymatic quenching (kinase/phosphatase inhibition) and degradation prevention.
Critical Procedures:
1. Rapid Collection & Quenching
- Cellular Samples: Aspirate media → Directly add ice-cold lysis buffer to culture vessels → Mechanically dislodge adherent cells on ice. For suspension cultures: flash-freeze pelleted cells (pre-chilled centrifuge) → Resuspend in lysis buffer.
- Tissues: Snap-freeze in liquid N₂ post-dissection → Pulverize under cryogenic conditions (mortar/pestle or cryomill) → Transfer powder to lysis buffer.
- Body Fluids (Plasma/Serum): Process immediately post-collection → Supplement with protease/phosphatase inhibitors → Clarify by centrifugation if required.
2. Lysis & Homogenization
- Lysis Buffer Composition:
- Chaotropes: 6-8M urea or guanidine HCl (protein denaturation/enzyme inactivation)
- Protease inhibitors: EDTA-free cocktail
- Phosphatase inhibitors: Comprehensive cocktail (e.g., NaF, Na₃VO₄, β-glycerophosphate, okadaic acid/calyculin A) targeting Ser/Thr and Tyr phosphatases
- Reducing agents: 5-10mM DTT or TCEP (disulfide bond reduction)
- Homogenization: Vigorous vortexing → Ice-cold probe sonication or mechanical homogenization → Verify complete lysis → Maintain ≤4°C throughout.
3. Protein Quantification
- Employ compatible assays:
- Urea-tolerant BCA method or Bradford assay
- Note: Precise quantification ensures downstream normalization and loading consistency.
4. Alkylation
- Treat with 20-50mM iodoacetamide (IAA) or chloroacetamide (CAM) → Incubate 30min darkness (room temperature) → Blocks reduced thiols to prevent:
- Disulfide reformation
- Artifactual modifications
5. Optional Purification (Recommended)
- Urea Dilution: Reduce [urea]<2M using Tris-HCl (pH~8) buffer to prevent carbamylation during digestion.
- Protein Precipitation: Methanol/chloroform or TCA/acetone → Removes salts/lipids/inhibitors → Concentrates proteins → Resuspend pellet in digestion buffer.
- Buffer Exchange: Centrifugal filtration (e.g., Amicon Ultra) → Transfer to optimal digestion buffer (e.g., 50mM NH₄HCO₃, pH 8.0).
Matters need attention
Input Requirements by Sample Type
- Minimum Input Thresholds: Establish species-specific minimums (cell counts, tissue mass, plasma volume). Note: Comprehensive profiling necessitates substantial inputs (>1mg total protein).
- Sample-Specific Benchmarks:
- Cultured cells: ≥10⁷ cells
- Solid tissues: ≥50mg wet weight
- Plasma/Serum: ≥100μL
2. Complexity Management
- Interfering Matrices (Tissue/Plasma):
- Implement enhanced purification for samples containing:
- High lipid/salt content
- Abundant carrier proteins (e.g., albumin)
- Recommended Mitigation: Immunodepletion • Organic extraction • Subcellular fractionation
- Implement enhanced purification for samples containing:
3. Rare/Precious Sample Protocols
- Micro-Scale Strategies (e.g., LCM, Primary Cells):
| Approach | Implementation | Limitations |
|---|---|---|
| Nano-volume lysis | ≤20μL buffers | Reduced protein recovery |
| Microfluidic enrichment | Chip-based phosphopeptide isolation | Throughput constraints |
| High-sensitivity MS | 30μm columns • 20nL/min flows | Restricted identification depth |
- Performance Expectations: Anticipate 30-50% reduced coverage versus conventional inputs
4. Detergent Compatibility
- Ionic Detergent Warnings:
- Strong ionic detergents (e.g., SDS) cause severe LC-MS interference through: Chromatographic artifacts, Ion suppression
- Mitigation Protocol: Sequential precipitations (e.g., acetone/methanol-chloroform) → Filtration columns (30kDa MWCO) → Confirm removal via BCA/blank MS runs
5. Reduction/Alkylation Tradeoffs
- Denaturant-Induced Modifications:
| Denaturant | Risk | Mechanism |
|---|---|---|
| Urea (>2M) | Protein carbamoylation | Isocyanic acid formation |
| IAA | Carboxymethylation | Alkylation side reactions |
- Optimized Protocol:
- Reduction: 5mM TCEP (30min, 25°C)
- Alkylation: 10mM CAM (20min, 4°C darkness) → Minimizes side reactions
6. Phosphatase Inhibitor Efficacy
- Inhibitor-Specific Considerations:
| Phosphatase Class | Effective Inhibitors | Critical Limitations |
|---|---|---|
| Ser/Thr | Okadaic acid, Calyculin A | pH instability (<8.0) |
| Tyr | Sodium orthovanadate | EDTA chelation → Inactivation |
- Best Practice: Freshly prepare inhibitor cocktails • Avoid EDTA-containing buffers • Validate activity via colorimetric assays
Protein Digestion: Complexity Reduction for Analysis
Objective: Generate peptide fragments optimized for LC-MS/MS characterization.
Enzyme Selection Rationale:
- Trypsin (Preferred): Cleaves C-terminal to lysine and arginine residues. Produces peptides with:
- Ideal length range (typically 7-25 aa)
- Favorable ionization properties (C-terminal basicity)
- Enhanced phosphosite retention during MS fragmentation
Standard Protocol:
- Enzyme-to-Protein Ratio: 1:20–1:50 (w/w); sample-dependent optimization recommended
- Reaction Conditions: 37°C incubation for 4–18 hours (overnight standard) at pH 8.0
- Termination:
- Acidify with formic acid (FA) or trifluoroacetic acid (TFA) to pH < 3.0
- Clarify by centrifugation → Remove precipitated debris
Alternative Enzymatic Strategies:
- Lys-C Application:
- Cleaves exclusively N-terminal to lysine
- Employed either:
- a) Solely for challenging samples
- b) Sequentially with trypsin (Lys-C digestion precedes trypsin) → Improved proteolytic efficiency for refractory proteins
Critical Enzymatic Digestion Considerations
1. Trypsin Activity Validation
- New Lot Verification: Assess enzymatic competence via standardized assays (e.g., BAPNA hydrolysis kinetics or BSA digestion efficiency tests) prior to sample processing.
2. Digestion Parameter Optimization
- Time/Temperature Tradeoffs: Extended incubation (>18h) or elevated temperatures (>37°C) increase risks of:
- Trypsin autolysis products
- Non-specific cleavages
- Refractory Sample Handling: Implement pre-digestion sonication (3×30s pulses, 30% amplitude) for insoluble pellets
3. Denaturant Management Protocol
- Urea Concentration Control (<2M):
| Method | Implementation | Verification |
|---|---|---|
| Dilution | Tris-HCl (pH 8.0) addition | Conductivity measurement |
| Buffer exchange | Zeba™ spin desalting columns | Ninhydrin test |
- Consequence of Neglect: Carbamylation artifacts → Peptide identification failures
Phosphopeptide Enrichment: Core to Phosphoproteomics
Objective: Selective isolation of substoichiometric phosphopeptides from dominant non-phosphorylated backgrounds.
Primary Enrichment Methodologies:
1. Titanium Dioxide (TiO₂) Chromatography
- Mechanism: Acidic phosphate-Ti⁴⁺ coordination (pH ≤ 2.5) enhanced by additives (DHB/lactic acid) suppressing non-specific binding.
- Workflow:
- Load acidified digest onto TiO₂ media (tip/column/magnetic beads)
- Wash with acidic loading buffer + organic solvent (e.g., acetonitrile)
- Elute phosphopeptides using alkaline buffer (e.g., NH₄OH, pH 10.5–11.5)
- Advantages: High Ser/Thr-phosphopeptide specificity, operational robustness
- Limitations: Reduced polyphosphorylated/phosphotyrosine (pY) recovery; Additive optimization critical
2. Immobilized Metal Affinity Chromatography (IMAC)
- Mechanism: Phosphate chelation by immobilized Fe³⁺/Ga³⁺/Ti⁴⁺ ions
- Workflow:
- Charge resin (Ni-NTA/IDA/Ti-IMAC) with metal ions
- Equilibrate with acidic buffer
- Load acidified digest
- Wash with acidified organic solvent
- Elute using alkaline or phosphate buffer
- Advantages: Superior polyphosphorylated/pY enrichment (Ga/Ti); High capacity
- Limitations: Non-specific binding (acidic/E/D-rich peptides); Complex resin preparation; Reduced reproducibility vs. TiO₂
Optimization Strategies:
- Sequential Enrichment: IMAC → TiO₂ workflow
- Hybrid Materials: Ti-IMAC resins
- Post-Enrichment Processing:
- Acidify + desalt via C₁₈ StageTips/mini-columns → Remove buffers/salts prior to MS
Critical Challenges in Phosphopeptide Enrichment
1. Non-Specific Binding Mitigation
- TiO₂ Optimization: Adjust DHB/lactic acid (20-50mM) in loading buffer → Suppress acidic peptide co-isolation
- IMAC Refinement:
| Parameter | Optimal Range |
|---|---|
| Acid concentration | 0.1-1% TFA |
| Organic solvent | 50-80% ACN |
| Wash volume | ≥10 column volumes |
2. Batch Consistency Management
- Material Variability:
- Different TiO₂/IMAC lots exhibit performance deviations → Implement:
- Single-batch usage for project continuity
- Pre-testing new batches with standard phosphopeptides
- Different TiO₂/IMAC lots exhibit performance deviations → Implement:
- QC Metric: Retention time shifts >2% warrant batch rejection
3. IMAC Metal Contamination Control
- Consequence:
- Free metal ions cause:
- MS signal suppression
- Ion source fouling
- Free metal ions cause:
- Protocol: Post-enrichment EDTA washes (5mM) → Triple rinsing with 0.1% FA/50% ACN
4. Phosphotyrosine (pY) Enrichment Specificity
- Inherent Challenges:
- pY abundance: 0.01-0.1% of pS/pT → Requires specialized approaches:
| Strategy | Implementation |
|---|---|
| pY-specific antibodies | Immune-affinity enrichment |
| Hybrid TiO₂-IMAC | Sequential enrichment |
- Expectation Setting: pY detection rates typically 30-50% lower than pS/pT
5. Elution Compatibility
- Silica Degradation Risk: Alkaline eluents (pH>11.5) dissolve silica-based matrices → Silicate MS contamination
- Safe Alternatives:
- 0.5% piperidine (pH 11.0)
- Phosphoric acid (500mM)
- Polymer-based TiO₂ tips
Liquid Chromatography (LC): Enhancing Separation Fidelity
Objective: Achieve temporal resolution of complex phosphopeptide mixtures prior to mass spectrometric introduction.
Methodology: Nanoflow Reversed-Phase LC (RP-LC)
- Chromatographic Configuration:
- Column: Fused silica capillary (75µm ID × 15-50cm) packed with C₁₈ stationary phase (1.7-3µm particles)
- Mobile Phase Composition:
- Solvent A: 0.1% formic acid in H₂O
- Solvent B: 0.1% formic acid in acetonitrile
- Separation Parameters:
- Gradient Profile:
- Linear increase from 2-5% B to 25-40% B over 90-120 minutes
- Note: Steeper gradients accelerate separation but compromise resolution
- Flow Rate: 200-400 nL/min (nanoflow regime)
- Rationale: Maximizes ionization efficiency via analyte concentration at nanoESI source
- Gradient Profile:
LC-MS/MS System Configuration & Performance Monitoring
1. Chromatographic Column Maintenance
- Column Conditioning: New columns require extensive equilibration (>10 column volumes) prior to sample analysis
- Operational Monitoring: Track backpressure deviations (±10%) and peak asymmetry changes (As >1.5 indicates degradation)
- Preventive Regeneration: Weekly flushing with 90% acetonitrile/0.1% FA → Monthly storage in >95% acetonitrile
2. Gradient Optimization Strategy
| Sample Complexity | Recommended Gradient | Peak Capacity Gain |
|---|---|---|
| Moderate | 90-min linear 5-35% B | ~400 |
| High | 180-min segmented gradient | ~700 |
| Ultra-complex | 300-min multi-step gradient | >900 |
3. Ion Source Contamination Control
- Risk Factor: Phosphoenriched samples carry residual matrix interferents →
- Source cone fouling
- Signal intensity decay
- Mitigation Schedule:
- Daily: Capillary wipe with methanol/water
- Weekly: Full source disassembly in 1:1:1 H₂O/MeOH/FA ultrasonication bath
4. Instrument Calibration Protocol
- Pre-Run Mandatory Calibration:
- Mass accuracy: ≤3 ppm error (LTQ tune mix)
- Resolution: ≥60,000 @ m/z 200
- Fragmentation energy: HCD optimization via Glu-1 fibrinopeptide
- Validation Frequency: Every 72 hours during continuous operation → Maintain<5% CV in QC standards
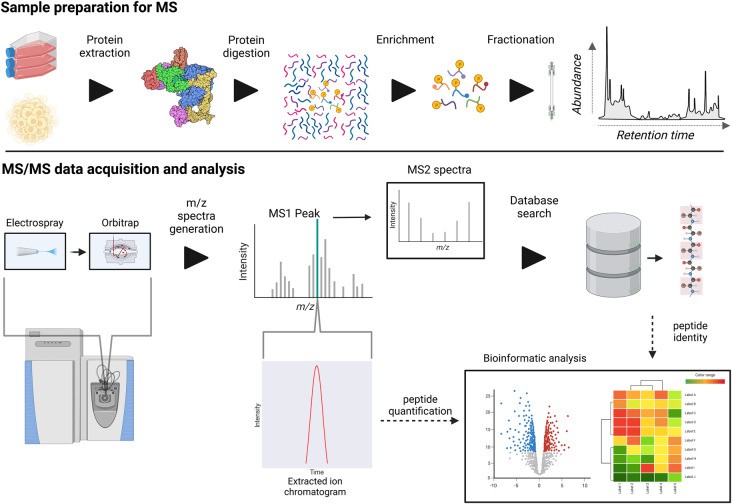 LC–MS/MS workflow for data-dependent analysis (DDA) phosphoproteomics (Higgins L et al., 2023)
LC–MS/MS workflow for data-dependent analysis (DDA) phosphoproteomics (Higgins L et al., 2023)
Select Service
Learn more
Mass Spectrometry: Detection and Identification Capabilities
Instrumentation: High-resolution/high-sensitivity tandem MS platforms, typically Orbitrap-based systems (e.g., Q-Exactive HF, Exploris, Lumos, Eclipse) or timsTOF instruments.
For more information on mass spectrometry and how to choose a platform, please refer to "Mass Spectrometry for Phosphoproteomics: Which Platform Is Best?".
Data Acquisition Strategies:
1. Data-Dependent Acquisition (DDA / "TopN")
- Workflow:
- Full MS1 scan (Resolution: 60-120k @ m/z 200) detects peptide ions
- Top N most intense precursors (N=10-20) selected for fragmentation per cycle
- Fragmentation: High-energy Collisional Dissociation (HCD) - Standard for phosphopeptides:
- Balanced backbone fragmentation
- Moderate phosphate group retention
- Advantages: Established workflows; Simplified data processing
- Limitations: Stochastic undersampling; Run-to-run variability; Abundance bias
2. Data-Independent Acquisition (DIA)
- Workflow:
- Full MS1 scan (High resolution)
- Sequential fragmentation of all precursors within consecutive m/z windows (e.g., 4-25 Da width)
- Advantages: Comprehensive precursor coverage; Enhanced quantitative reproducibility (multi-sample studies)
- Limitations: Complex data analysis requiring spectral libraries and specialized software (e.g., Spectronaut, DIA-NN)
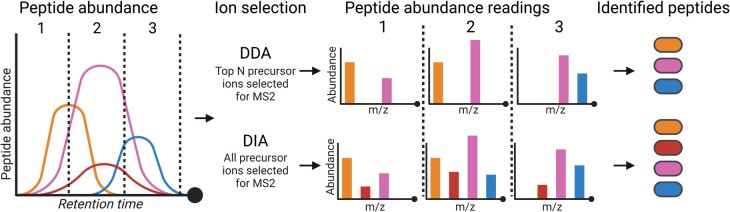 Overview of DIA and DDA approaches in proteomics (Higgins L et al., 2023)
Overview of DIA and DDA approaches in proteomics (Higgins L et al., 2023)
Phosphorylation-Specific Enhancements:
- Neutral Loss Triggering: Instruments configured to trigger MS³ upon detecting diagnostic neutral losses (-98 Da H₃PO₄ / -80 Da HPO₃) → Improved phosphosite localization confidence
- Multiplexed Fragmentation (e.g., SPS-MS³): Simultaneous isolation/fragmentation of multiple precursors → Reduced co-isolation interference → Enhanced TMT/iTRAQ quantitation accuracy
Data Analysis: Translating Spectra to Biological Insight
Objective: Identify phosphopeptides, localize modification sites, and quantify differential abundance across samples.
Core Analytical Workflow:
Raw Data Conversion:
- Utilize tools (e.g., ProteoWizard's MSConvert) to transform vendor-specific files into open formats (.mzML/.mzXML).
Database Searching (DDA):
- Software: Mascot, Sequest (via Proteome Discoverer), MaxQuant/Andromeda, MSFragger, Comet
- Input Parameters:
- MS/MS spectra
- Species-specific FASTA database
- Enzymatic specificity (Trypsin)
- Modifications:
- Fixed: Carbamidomethylation (C)
- Variable: Oxidation (M), Phosphorylation (S/T/Y)
- Output: Peptide-spectrum matches (PSMs) with localization scores
Phosphosite Localization:
- Tools: Ascore, PhosphRS, PTMProphet, LuciPHOr2, SLoMo
- Algorithmic Principle: Evaluate fragment ion evidence for each candidate phosphosite → Assign localization probability (≥75% confident; ≥95% high-confidence)
False Discovery Control:
- Implement target-decoy strategy with FDR thresholds (PSM/peptide/protein ≤1%)
Quantification Approaches:
- Label-free (LFQ): MaxLFQ algorithm (intensity-based) requiring retention time alignment and normalization
- Isobaric Tags (TMT/iTRAQ): Reporter ion intensity measurement (MS²/MS³-level) with inter-channel normalization
DIA-Specific Processing:
- Requirements: Project-specific spectral libraries (DDA-derived) or public repositories
- Software: Spectronaut, DIA-NN, Skyline, MaxDIA
- Workflow: Match DIA fragments to library spectra → Simultaneous identification/quantification with phosphosite scoring
Downstream Interpretation:
- Data Filtering: Localization probability >0.75; unique peptides
- Statistical Analysis: t-test/ANOVA for significant phosphosites (p<0.05; FDR-adjusted)
- Functional Mapping:
- Pathway enrichment (KEGG/Reactome/GO via STRING, IPA)
- Kinase-substrate networks (KSEA, PhosR)
- Visualization: Volcano plots; heatmaps; phosphoproteomic landscapes
How to use R language and tools to analyze data, please refer to "How to Analyze Phosphoproteomics Data with R and Bioinformatics Tools".
Data Analysis Complexities and Strategic Considerations
1. Database Selection Integrity
Employing accurate, species-specific protein databases that comprehensively include isoforms and splice variants is imperative. Public repositories may contain inaccuracies or omissions, necessitating rigorous cross-referencing with curated resources.
2. Variable Modification Search Optimization
| Parameter | Impact | Balancing Strategy |
|---|---|---|
| Added modifications | Exponential search space expansion | Limit to biologically relevant |
| (e.g., oxidation, acetylation) | Increased computational time | modifications only |
| Reduced identification sensitivity |
3. Phosphosite Localization Interpretation
- Probability Score Limitations: Even 99% localization confidence cannot ensure absolute accuracy for adjacent Ser/Thr clusters.
- Validation Protocol:
- Integrate MS/MS fragment evidence with: Sequence motif conservation • Phosphorylation site databases (PhosphoSitePlus) • Pathway context
4. Quantitative Normalization Framework
| Method | Application | Inherent Limitations |
|---|---|---|
| Total Protein Intensity | Global normalization | Assumes constant proteome |
| Median Abundance Scaling | Label-free experiments | Sensitive to composition shifts |
| Reference Phosphopeptides | Phosphoproteome-specific | Requires stable phosphospecies |
- DIA Criticality: Normalization is paramount for cross-run comparisons in data-independent acquisition.
5. Missing Value Management
- Handling Approaches:
| Strategy | Implementation | Statistical Impact |
|---|---|---|
| Zero Imputation | Assign null values as zero | Inflation of type I errors |
| Detection Limit Imputation | Replace with LOD/2 values | Conservative bias introduction |
| Complete Case Analysis | Exclude missing observations | Reduced statistical power |
- Best Practice: Apply multiple imputation with Markov Chain Monte Carlo (MCMC) modeling for minimal bias.
Quality Assurance & Validation: Ensuring Data Integrity
Workflow Monitoring Measures:
- Replication Strategy: Incorporate biological/technical replicates to assess variability
- Positive Controls: Spike known phosphopeptides (e.g., Sigma UPS2 mix) → Monitor enrichment efficiency + instrument sensitivity
- Negative Controls: Analyze non-enriched digests → Evaluate enrichment specificity
- System Suitability: Inject solvent blanks between samples → Detect carryover
Data QC Metrics:
| Parameter | Benchmark | Purpose |
|---|---|---|
| Enrichment Efficiency | >70% phosphopeptides/total IDs | Specificity assessment |
| Phosphosite Localization | >75% phosphopeptides with ≥75% probability | Modification confidence |
| Technical Reproducibility | Pearson's r >0.8 (LFQ intensities) | Measurement precision |
| MS Instrument Performance | MS/MS acquisition rate; mass accuracy<5ppm | System stability verification |
Orthogonal Validation:
- Immunoblotting: Phospho-specific antibodies → Confirm key phosphoprotein/phosphosite alterations
- Targeted MS (PRM): Quantify critical phosphopeptides across samples → High-specificity verification
Critical Success Factors for CRO Collaborations:
- Sample Integrity: Degraded samples or inadequate phosphatase inhibition compromise data reliability → Disclose collection/processing details
- Biological Objectives: Define exploratory/targeted goals → Determine experimental design (replicates, labeling, depth)
- Method Transparency: Clarify enrichment methodology (TiO₂/IMAC/hybrid) and rationale → Anticipate technical limitations
- Throughput-Resolution Tradeoff: Prioritize: Deep coverage (long gradients/replicates) vs. high-throughput analysis
- Analytical Specifications:
- Confirm deliverables:
- Identification depth
- Quantitation rigor (statistical thresholds)
- Pathway analysis inclusion
- Localization confidence criteria
- Confirm deliverables:
- Data Deliverables:
- Specify formats: Raw outputs (Proteome Discoverer/MaxQuant) → Processed results → DIA spectral libraries
- Proactive Communication:
- Disclose sample metadata (type, quantity, lysis buffer, issues)
- Maintain iterative dialogue throughout the project
conclusion
Achieving high-quality, biologically significant data demands rigorous command of the end-to-end analytical process—from safeguarding labile phosphospecies during sample preparation through optimized digestion, targeted enrichment, high-resolution separation, sensitive mass spectrometry, and advanced bioinformatic interpretation. For laboratory specialists, meticulous execution at every phase remains non-negotiable. For CRO clients, comprehending this pipeline enables informed experimental design decisions, appropriate sample submission practices, and nuanced interpretation of complex datasets. By internalizing the methodological intricacies and critical control points detailed herein, both stakeholders can harness phosphoproteomics to decipher cellular signaling dynamics with unprecedented resolution.
References
- Wu X, Liu YK, Iliuk AB, Tao WA. "Mass spectrometry-based phosphoproteomics in clinical applications." Trends Analyt Chem. 2023 Jun;163:117066. doi: 10.1016/j.trac.2023.117066
- Higgins L, Gerdes H, Cutillas PR. "Principles of phosphoproteomics and applications in cancer research." Biochem J. 2023 Mar 31;480(6):403-420. doi: 10.1042/BCJ20220220
- Muneer G, Chen CS, Chen YJ. "Advancements in Global Phosphoproteomics Profiling: Overcoming Challenges in Sensitivity and Quantification." Proteomics. 2025 Jan;25(1-2):e202400087. doi: 10.1002/pmic.202400087











