Phosphoproteomics provides essential access to deciphering cellular signaling networks, disease pathways, and therapeutic targets. However, this approach presents significant methodological challenges: phosphorylation events are inherently transient, occur at low stoichiometry, and exhibit marked susceptibility to degradation. Crucially, the integrity of sample preparation critically influences the success of downstream mass spectrometry and frequently determines overall project viability. This article delivers actionable, step-by-step protocols tailored for laboratory researchers and CRO specialists to optimize experimental workflows.
Core concept: speed, low temperature and strong inhibition
Successful phosphorylation analysis demands adherence to three core principles: rapid processing, sustained low temperatures, and comprehensive enzymatic blockade.
1. Universal Precooling
All experimental equipment and reagents must undergo precooling to minimize undesired enzymatic activity. Key procedures involve:
- Icing Work Surfaces: Maintain centrifuge tubes, pipette tips, and work areas on ice to prevent temperature-induced enzyme activation.
- Chilled Centrifuge Rotors: Ensure rotors are pre-cooled, as elevated temperatures accelerate enzymatic reactions during centrifugation.
- Precooled Transfer Tools: Centrifuge tubes and pipette tips must be chilled to avoid sample exposure to ambient conditions.
2. Expedited Sample Handling
Processing delays risk phosphorylation signal loss or alteration. Maximize information retention through immediate and efficient action:
- Tissue Processing: Initiate lysis instantly upon tissue isolation to limit ambient temperature exposure.
- Cell Treatment: Add lysis buffer right after cell disruption to prevent endogenous enzymatic processes.
3. Comprehensive Inhibition
Ensuring result reliability necessitates blocking both phosphatase and protease activity via broad-spectrum inhibitors:
- Phosphatase Blockade: Employ inhibitors like sodium molybdate or sodium fluoride to effectively suppress diverse phosphatase types.
- Protease Suppression: Prevent protein degradation during lysis using appropriate inhibitors (e.g., PMSF, EDTA, Leupeptin).
4. Continuous Temperature Control
Maintain sub-ambient temperatures throughout critical steps (e.g., on ice or at 4°C) to suppress enzymatic activity:
- Lysis: Perform cell or tissue disruption swiftly on ice to prevent enzyme over-activation.
- Precipitation: Conduct protein precipitation under chilled conditions to minimize degradation.
- Storage: Immediately preserve samples at -80°C or in liquid nitrogen post-experiment for long-term phosphorylation stability.
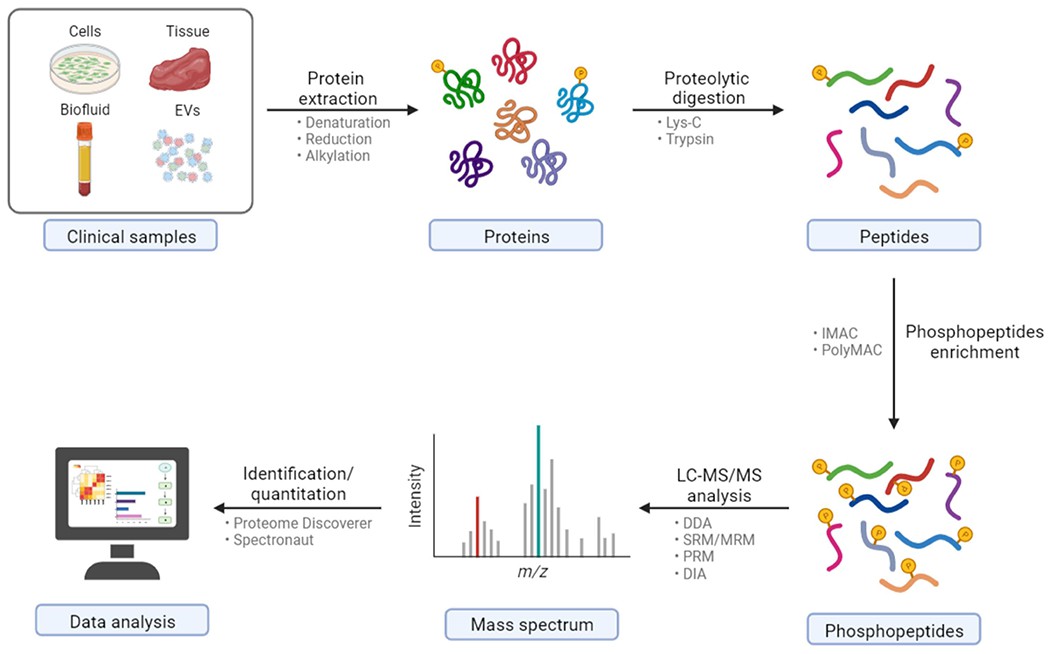 General workflow of clinical sample preparation for bottom-up phosphoproteomics via LC-MS/MS (Wu X et al., 2023)
General workflow of clinical sample preparation for bottom-up phosphoproteomics via LC-MS/MS (Wu X et al., 2023)
Critical Procedural Guidelines and Optimized Methodologies
1. Specimen Pretreatment Protocols
Tissue Processing:
- Rapid Cryopreservation: Immediately flash-freeze excised tissues in liquid nitrogen (≤30 seconds post-extraction). Dry ice methods are discouraged due to suboptimal cooling kinetics.
- Cryogenic Fragmentation: Perform grinding/ball milling within liquid nitrogen using precooled equipment to prevent thawing.
- Concurrent Lysis: Introduce chilled lysis buffer (10× sample volume) during disruption to halt enzymatic activity immediately.
Cell Processing (Adherent/Suspension):
- Ice-Cold Washes: Utilize PBS containing phosphatase inhibitors for rapid rinsing. Post-centrifugation (500g, 4°C, 5 min), remove supernatant completely.
- Non-Mechanical Harvesting: Directly add lysis buffer to culture vessels, avoiding scrapers that induce physical stress.
- Trypsin Neutralization: When enzymatically detaching cells, extensively wash with inhibitor-supplemented PBS to eliminate residual trypsin activity.
2. Lysate Formulation and Implementation
Standard Composition:
- Denaturants: 8M urea or 6-8M thiourea to unfold protein structures
- Detergents: 2% CHAPS or 4% SDS (preferred for insoluble targets)
- Buffer System: 50mM Tris-HCl (pH 8.0)
- Ionic Strength: 75mM NaCl
- Enzyme Suppression:
- Phosphatase inhibitors (e.g., PhosSTOP™, NaF/Na3VO4)
- Protease inhibitors (e.g., EDTA-free cocktail)
Execution Guidelines:
- Buffer Freshness: Prepare inhibitor-containing lysate immediately before use. Aliquot and freeze at -80°C; thaw on ice when needed.
- Volume Optimization: Maintain immersion ratios: 1:10 (w/v) for tissues, 1×10⁷ cells/mL for cultures.
- Mechanical Disruption: Employ cryo-sonication (low-power pulsed mode) or vortexing with protease inhibitors. Prevent temperature elevation during processing.
- Clarification: Centrifuge lysates (14,000-16,000 g, 4°C, 15-20 min). Carefully transfer supernatant without disturbing pelleted debris.
Methodological Synthesis and Enhancements
- Sustain cryogenic conditions throughout pretreatment to prevent protein deterioration
- Minimize freeze-thaw cycles through aliquot-based lysate storage
- Implement appropriate physical disruption to maximize lysis efficiency
- Prompt post-lysis centrifugation ensures analyte purity for downstream applications
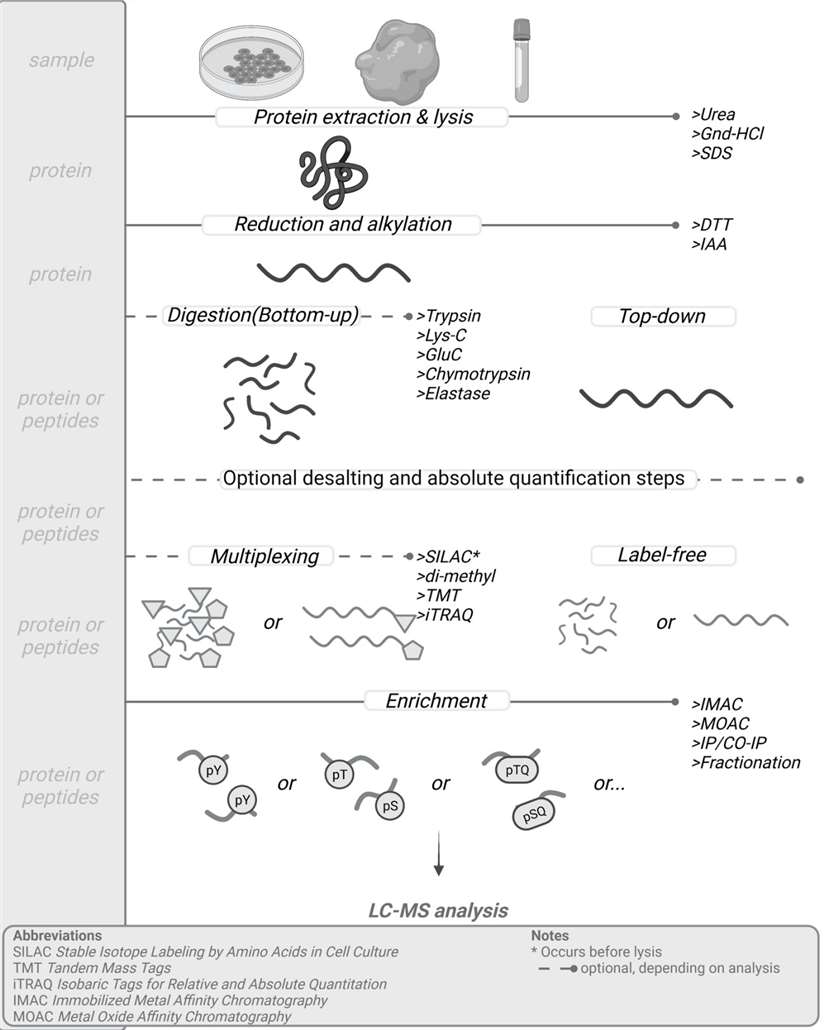 General sample preparation protocol for mass-spec based phosphoproteomic analysis (Gerritsen JS et al., 2021)
General sample preparation protocol for mass-spec based phosphoproteomic analysis (Gerritsen JS et al., 2021)
3. Protein Precipitation and Reconstitution Protocol
Rationale for Precipitation
Protein precipitation eliminates contaminants (salts, detergents, lipids) that impede downstream enzymatic digestion, enrichment, and analytical procedures. This process alters solvation conditions to reduce protein solubility, facilitating selective isolation.
Cryo-Acetone Precipitation Methodology
Procedure:
- Precipitation Initiation: Add 4 volumes of pre-chilled acetone (-20°C) to lysate. Acetone disrupts protein-solvent interactions, inducing aggregation.
- Incubation: Maintain at -20°C for ≥4 hours (overnight recommended) for complete protein separation.
- Pellet Isolation: Centrifuge (14,000g, 4°C, 15 min). Exercise caution during supernatant removal to preserve pellets.
- Contaminant Removal: Wash pellets twice with chilled 90% acetone using gentle aspiration.
- Solvent Evaporation: Air-dry pellets 10-15 minutes. Excessive drying compromises redissolution efficiency.
- Protein Reconstitution: Resuspend in strong denaturant buffer (e.g., 8M urea/50mM TEAB) using vortexing or brief sonication.
Critical Parameters:
- Acetone Temperature: Strict -20°C maintenance maximizes precipitation efficiency.
- Drying Control: Complete acetone removal prevents interference with subsequent steps.
- Redissolution Validation: Ensure no particulate residues remain before proceeding.
4. Protein Quantitation Analysis
Accurate concentration determination is essential for downstream processing. Select methods based on buffer compatibility:
BCA Assay (Denaturant-Tolerant):
Measures Cu²⁺ reduction by peptide bonds forming a colorimetric complex. Exhibits moderate resistance to denaturants (validated for ≤5M urea). Quantify via standard curve using BSA references.
Bradford Assay (Limited Compatibility):
Detects Coomassie dye shift upon protein binding. Avoid with SDS-containing samples (>0.1%) or strong chaotropes due to signal interference.
Optimal Practices:
- Calibration: Generate fresh standard curves for each experiment.
- Homogenization: Vortex samples thoroughly before measurement.
- Replication: Perform triplicate readings, particularly for dilute samples.
5. Disulfide Bond Management and Proteolytic Digestion Protocol
Disulfide Reduction
- Objective: Cleave disulfide bonds to generate free thiol groups, enabling subsequent alkylation. This is critical for structural analysis of disulfide-containing proteins.
- Procedure:
- Reagent: Dithiothreitol (DTT, 5–10 mM final concentration)
- Conditions: 20–60 min at RT or 56°C (elevated temperatures risk non-specific modifications)
- Stability: Minimize air exposure; prepare fresh solutions and maintain chilled
Thiol Alkylation
- Objective: Permanently cap reduced cysteine residues to prevent re-oxidation.
- Procedure:
- Reagents: Iodoacetamide (IAM, 20–40 mM) or chloroacetamide (55 mM)
- Conditions: 30–45 min at RT in darkness (alkylators are photosensitive)
- Urea Precautions: Avoid extended reactions in urea buffers to prevent carbamylation artifacts
Proteolytic Digestion
- Objective: Generate peptide fragments suitable for mass spectrometry.
- Pre-Digestion Requirements:
- Urea Dilution: Reduce urea concentration to<2M via:
- Buffer dilution (50mM TEAB/ABC)
- Desalting chromatography
- Digestion Protocol:
- Enzyme: Sequencing-grade trypsin (cleaves C-terminal to Lys/Arg)
- Ratio: 1:25–1:50 (w/w, enzyme:protein)
- Conditions: 37°C for 16–18 hr
- Termination: Add TFA (0.5–1%) or formic acid (2–5%)
- Urea Dilution: Reduce urea concentration to<2M via:
Critical Reaction Parameters
| Parameter | Requirement | Rationale |
|---|---|---|
| Temperature | Strict maintenance | Prevents non-specific modifications |
| Time Control | Step-specific limits | Avoids side reactions |
| Light Exposure | Alkylation in dark | Preserves reagent efficacy |
| Inhibitors | Added for long incubations | Minimizes autoproteolysis |
6. Phosphopeptide Enrichment: Critical Methodologies
Rationale for Enrichment
Phosphopeptides typically represent<0.1% of total peptide populations, necessitating high-efficiency isolation prior to downstream analysis.
Comparative Enrichment Matrix
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| TiO₂ | Metal oxide affinity |
|
|
| IMAC | Fe³⁺/Ga³⁺ metal coordination |
|
|
| Ti-IMAC | Titanium-immobilized affinity |
|
|
Optimization Framework
- Acidic Environment Control
- Loading/wash buffers must contain:
- ≥80% acetonitrile
- >1% TFA (or alternatives: 6% DHB/2,5-dihydroxybenzoic acid)
- Sample Pretreatment
- Implement rigorous desalting for SDS-containing lysates
- Precipitate high-salt specimens before enrichment
- Low-Binding Surfaces: Use polymer-coated tubes and tips to minimize surface adsorption
- Enrichment Repetition: Employ serial enrichment or high-capacity resins for complex matrices
- Process Validation: Include phosphopeptide standards in each batch for QC assessment
7. Desalting and Sample Preservation
Desalting Methodology
C18 solid-phase extraction (StageTip® or centrifugal columns) performs essential buffer exchange and salt removal, enhancing MS signal intensity and analytical stability.
Storage Protocols
| Duration | Conditions | Format |
|---|---|---|
| Short-term | 4°C in 0.1% formic acid/water | Aqueous solution |
| Long-term | -80°C frozen OR Vacuum-concentrated | Lyophilized powder |
Quality Control Framework
1. Pre-Analytical Electrophoretic Assessment
Objective: Validate protein extract integrity through preliminary electrophoretic separation and immunoblotting prior to formal analysis.
| Method | Application | Detection Capability |
|---|---|---|
| Coomassie | Initial screening | High-contrast visualization (limited sensitivity) |
| Silver Stain | Low-abundance targets | Enhanced detection threshold (≥100× Coomassie) |
| Western Blot | Phosphorylation verification | Pan-phospho-Ser/Thr/Tyr antibody validation |
Critical Implementation:
- Perform whole-protein electrophoresis to confirm distribution uniformity and structural integrity
- Utilize pan-phospho antibodies to establish phosphorylation preservation efficacy
2. Pre-MS Peptide Evaluation
Objective: Optimize mass spectrometry conditions through preliminary abundance assessment.
Protocol:
- Quantitation: Determine peptide concentration post-digestion using UV spectrophotometry (e.g., Nanodrop™)
- Prescanning: Conduct rapid MS reconnaissance to:
- Verify phosphopeptide signal intensity
- Identify potential ion suppression issues
- Adjust injection volumes for optimal detection
3. Isotopic Standard Integration
Objective: Monitor procedural recovery efficiency using reference phosphopeptides.
Workflow:
- Spike known quantities of synthetic isotope-labeled phosphopeptides during digestion
- Track standards through extraction → digestion → enrichment → analysis
- Calculate stepwise recovery rates (acceptance: >60% overall)
- Troubleshooting: Suboptimal recovery indicates sample loss at specific stages
4. Pre-Interpretation QC Metrics
Objective: Ensure analytical robustness before bioinformatic processing.
Critical Checks:
- Spectral Filtering: Eliminate spectra with:
- Signal-to-noise ratio<25
- Precursor mass error >10 ppm
- Quantitative Validation:
- Confirm reproducibility across technical replicates (CV<15%)
- Verify linearity via standard curves (R² >0.98)
- Phosphosite Confirmation: Require:
- Localization probability >0.75 (e.g., Ascore, PTM-Score)
- MS/MS fragmentation coverage ≥5 consecutive b/y ions
Essential Materials for Phosphoproteomic Workflows
Reagent Specifications
- Protein Denaturation:
- Urea (ultrapure grade) or thiourea
- Chaotropic detergents: SDS (electrophoresis grade) or CHAPS
- Enzyme Suppression:
- Phosphatase/protease inhibitor cocktails (e.g., cOmplete™, PhosSTOP™)
- Proteolytic Agents:
- Trypsin (sequencing-grade, e.g., Promega)
- Disulfide Management:
- Reducing agent: DL-dithiothreitol (DTT)
- Alkylating agents: Iodoacetamide (IAA) or chloroacetamide (CAA)
- Solvent Systems:
- HPLC-grade acetonitrile (ACN) and acetone
- Ionization-compatible acids: Trifluoroacetic acid (TFA), formic acid (FA), acetic acid
- Enrichment/Desalting:
- Phosphopeptide isolation kits (TiO₂, IMAC, Ti-IMAC)
- C18 stationary phase media (columns or StageTip® formats)
Instrumentation Requirements
| Category | Critical Equipment |
|---|---|
| Temperature Control | Pre-cooled high-speed centrifuges |
| Sample Processing | Vortex mixers, probe sonicators |
| Incubation | Thermostatic shakers/water baths |
| Concentration | Vacuum centrifugal concentrators |
| Monitoring | Precision pH meters |
| Storage | -80°C freezers/liquid N₂ dewars |
Critical Consumables
- Low-protein-binding microcentrifuge tubes and pipette tips
- Cryogenic disruption tools: Liquid nitrogen-chilled mortars, ball mill beads
- Sterile 0.22 μm PVDF membrane filters (buffer sterilization)
Select Service
Learn more
Pit Avoidance Guide
1. Inhibitor Efficacy
Issue: Compromised phosphorylation signals occur when samples aren't maintained at low temperatures throughout processing or when expired inhibitors are utilized.
Solution:
- Strict cryopreservation: Keep all samples and associated reagents on ice or within an ultra-low temperature environment during every experimental step to prevent degradation of proteins and their phosphorylation sites.
- Verify inhibitor activity: Employ freshly prepared protease and phosphatase inhibitors, routinely checking expiration dates to exclude outdated reagents.
2. SDS Contamination
Issue: Signal loss or interference during phosphopeptide enrichment can stem from SDS residues persisting within the sample.
Solution:
- Rigorous desalting: Prior to enzymatic digestion, implement desalting techniques (e.g., C18 columns, dialysis) for SDS removal. Ethanol or isopropanol precipitation at varied concentrations offers an additional removal step.
- Confirm SDS elimination: Utilize UV absorbance measurements to quantify and ensure complete SDS removal from samples.
3. Incomplete Proteolysis
Issue: Suboptimal enzyme-to-substrate ratios or inadequate digestion duration yield residual large peptide fragments, hampering downstream analysis.
Solution:
- Optimize digestion parameters: Tailor conditions (temperature, duration, enzyme concentration) to the protein's characteristics. Standard protocols often use 37°C with a 1:50 to 1:100 (enzyme:protein) ratio.
- Assess digestion completeness: Post-digestion, employ SDS-PAGE or preliminary mass spectrometry to confirm sufficient breakdown of large peptides.
4. Suboptimal Phosphopeptide Recovery
Issue: Factors like inadequate enrichment buffer acidity, sample overloading, or peptide adsorption to tube surfaces diminish phosphopeptide yields.
Solution:
- Optimize buffer pH: Maintain phosphopeptide enrichment buffer between pH 2.0 and 3.0 to enhance adsorption and separation efficiency.
- Prevent column overloading: Excessive sample amounts dilute phosphopeptides and impede complete binding during enrichment.
- Minimize surface adsorption: Utilize low-binding tubes (e.g., coated plastic, PTFE) to reduce peptide loss.
5. Inadequate Reduction and Alkylation
Issue: Poor cleavage of disulfide bonds due to insufficient reduction/alkylation compromises digestion efficiency and modification site identification.
Solution:
- Refine reaction conditions: Apply reducing agents (DTT, TCEP) at adequate concentrations for sufficient time (typically 30-60 min), followed promptly by alkylating agents (IAA, IAM).
- Monitor reaction efficacy: Verify complete disulfide bond cleavage using SDS-PAGE or mass spectrometric assessment.
6. Incomplete Sample Cleanup
Issue: Excessive impurities (endogenous proteins, contaminants) obscure mass spectrometry data, hindering protein detection and quantification.
Solution:
- Implement multi-stage purification: Apply sequential techniques (affinity chromatography, centrifugation, precipitation) to deplete impurities.
- Integrate chromatography: Pre-enrichment, utilize reverse-phase HPLC or similar methods for further phosphopeptide purification, boosting detection sensitivity.
7. Data Analysis Inaccuracies
Issue: Misidentification or omission of phosphorylation sites can arise from database matching errors or unoptimized search parameters during data analysis.
Solution:
- Employ appropriate tools: Utilize relevant databases (e.g., UniProt) and current search algorithms (e.g., SEQUEST, Mascot) for analysis.
- Conduct multi-faceted validation: Analyze datasets with multiple software platforms to confirm consistency. Implement stringent quality control, such as FDR (False Discovery Rate) analysis, to minimize false positives.
8. Sample Processing Variability
Issue: Inconsistent handling across different sample batches compromises experimental result comparability.
Solution:
- Standardize protocols: Adhere to a strictly defined, identical operational workflow for every sample batch.
- Perform inter-batch controls: Include standardized control samples within each experimental run to verify consistent processing conditions across batches.
To learn more about the workflow, please refer to "Phosphoproteomics Workflow Explained: From Sample to Data".
For more information on mass spectrometry and how to choose a platform, please refer to "Mass Spectrometry for Phosphoproteomics: Which Platform Is Best?".
People Also Ask
Which technique is often used to enrich phosphopeptides prior to mass spectrometry analysis in phosphoproteomics?
Immobilized metal affinity chromatography. Currently, IMAC is one of the most widely used enrichment techniques for phosphopeptides prior to MS analysis. It enriches phosphopeptides by utilizing the affinity of the positively charged metal ions for negatively charged phosphate.
References
- Wu X, Liu YK, Iliuk AB, Tao WA. "Mass spectrometry-based phosphoproteomics in clinical applications." Trends Analyt Chem. 2023 Jun;163:117066. doi: 10.1016/j.trac.2023.117066
- Gerritsen JS, White FM. "Phosphoproteomics: a valuable tool for uncovering molecular signaling in cancer cells." Expert Rev Proteomics. 2021 Aug;18(8):661-674. doi: 10.1080/14789450.2021.1976152
- Sampadi B, Mullenders LHF, Vrieling H. "Phosphoproteomics Sample Preparation Impacts Biological Interpretation of Phosphorylation Signaling Outcomes." Cells. 2021 Dec 3;10(12):3407. doi: 10.3390/cells10123407











