Sulfatides Analysis Service
Creative Proteomics specializes in advanced lipidomics, offering high-sensitivity sulfatide quantification using Agilent 6495C and 1290 UHPLC platforms. We detect over 70 sulfatide species and related metabolites, enabling researchers to uncover lipid pathway dynamics, membrane biology, and molecular mechanisms with speed and precision.
Submit Your Request Now
×
- What We Provide
- Advantage
- Workflow
- Technology Platforms
- Sample Requirements
- FAQ
- Publication
What are Sulfatide?
Sulfatides are sulfated galactosylceramides—negatively charged glycosphingolipids—predominantly found in the nervous system and myelin sheaths, but also in kidneys, pancreas, and various epithelial tissues. These lipids play essential roles in membrane stability, cell adhesion, immune modulation, and signaling pathways. Due to their complexity and low abundance, precise quantification and structural identification of sulfatides require high-resolution analytical techniques.
Sulfatide Analysis Service Offered by Creative Proteomics
- Comprehensive Sulfatide Profiling: In-depth qualitative and quantitative analysis of sulfatide species, including hydroxylated and non-hydroxylated forms across various carbon chain lengths and saturation levels.
- Targeted Quantification Using Internal Standards: Absolute quantification of sulfatides utilizing isotope-labeled internal standards for unmatched accuracy and reproducibility.
- Structural Characterization by MS/MS: Advanced tandem mass spectrometry techniques for detailed elucidation of sulfatide molecular structures and isomer differentiation.
- Comparative Lipidomics Studies: Customized comparative analyses across biological conditions (e.g., developmental stages, diet, genotype, environment) to uncover sulfatide-level variations.
- Hydroxylation Status & Unsaturation Analysis: Determination of specific hydroxylation and unsaturation patterns that affect sulfatide bioactivity and membrane interactions.
- Tissue-Specific Sulfatide Mapping: Profiling of sulfatides in specialized tissues such as brain, liver, kidney, or nervous system components, based on client requirements.
- Quantitative Time-Course Analysis: Monitoring sulfatide concentration changes over time in longitudinal or time-course experimental designs.
- Data Visualization & Bioinformatics Reporting: Delivery of high-quality visual outputs (PCA, heatmaps, clustering) along with pathway-based interpretation to support publication or internal decision-making.
List of Detected Sulfatide and Related Metabolites
| Category | Representative Detected Molecules | Associated Metabolic Pathways |
|---|---|---|
| Sulfatides (Galactosylceramide Sulfates) | ST(d18:1/16:0), ST(d18:1/18:0), ST(d18:1/24:0), ST(d18:1/24:1) | Sphingolipid metabolism |
| Hydroxylated Sulfatides | ST(d18:1/18:0-OH), ST(d18:1/24:0-OH), ST(d18:1/24:1-OH) | Myelin sheath biosynthesis |
| Lactosylsulfatides | Lactosylceramide sulfate (d18:1/16:0), (d18:1/24:1) | Glycosphingolipid biosynthesis |
| Glucosylceramide Sulfates | GlcCer-S(d18:1/18:1), GlcCer-S(d18:1/20:0) | Glucosylceramide metabolism |
| Ceramides (precursors) | Cer(d18:1/16:0), Cer(d18:1/18:0), Cer(d18:1/24:0) | Ceramide biosynthesis pathway |
| Sphingoid Bases | Sphingosine (d18:1), Sphinganine (d18:0), Phytosphingosine (t18:0) | Sphingosine/Sphinganine degradation |
| Sulfate Donors | 3′-Phosphoadenosine-5′-phosphosulfate (PAPS) | Sulfation pathway |
| Sphingomyelins (related class) | SM(d18:1/16:0), SM(d18:1/24:1) | Sphingomyelin cycle |
| Fatty Acids (side chains) | Palmitic acid (C16:0), Stearic acid (C18:0), Lignoceric acid (C24:0) | Fatty acid elongation & desaturation |
| Sulfated Glycosphingolipid Intermediates | Monohexosylceramide-Sulfate, Dihexosylceramide-Sulfate | Glycosphingolipid sulfation pathway |
Advantages of Sulfatide Assay
- Ultra-High Sensitivity: Lower limit of quantification (LLOQ) reaches 0.1 ng/mL, enabling detection of ultra-trace sulfatides in plasma, tissue, and cell extracts.
- Exceptional Quantitative Precision: Method validation demonstrates intra-batch CV ≤ 8% and inter-batch CV ≤ 12% across all major sulfatide classes.
- High Analytical Throughput: With a runtime of ≤ 12 minutes per sample, our platform supports high-throughput analysis for >100 samples per batch.
- Broad Coverage: Over 70 sulfatide species detectable, including saturated, unsaturated, and hydroxylated forms across various acyl chain lengths (C16:0 to C26:1).
- Excellent Recovery Efficiency: Optimized extraction yields a recovery rate ≥ 85%, ensuring robust quantification even from low-input samples.
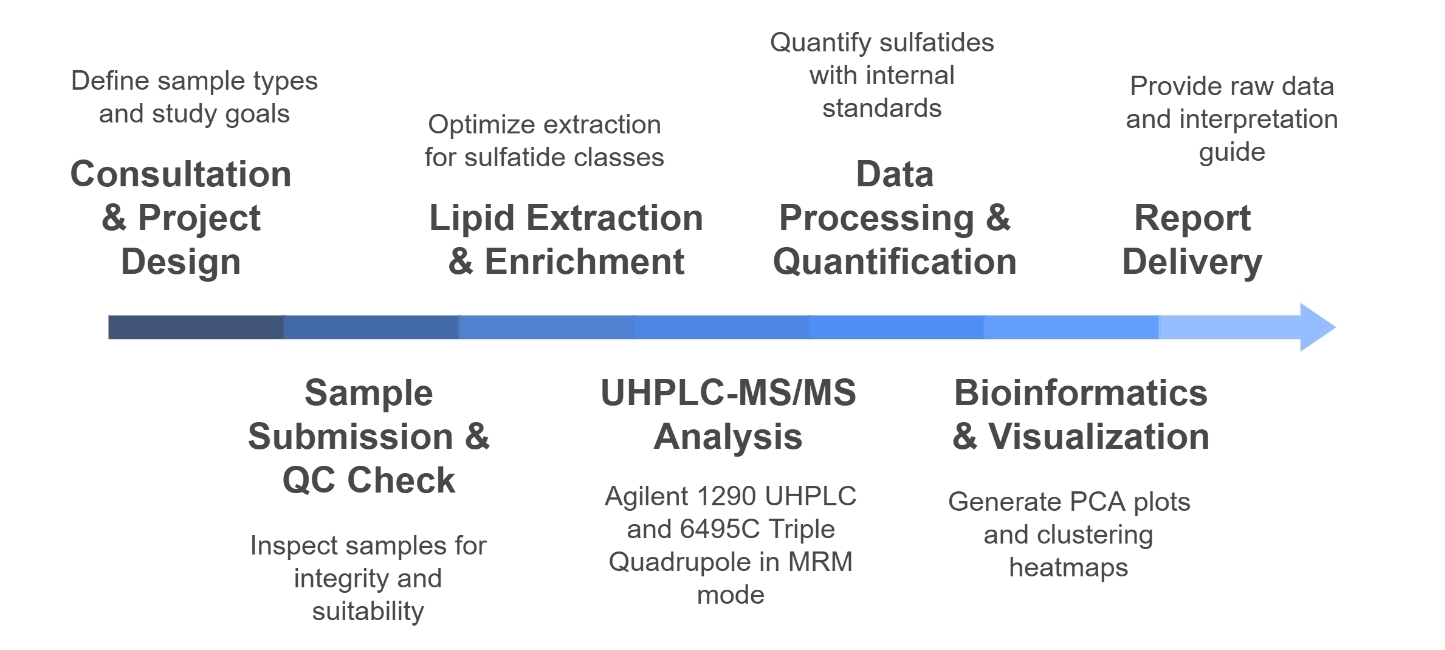
Workflow for Sulfatide Analysis Service
Technology Platform for Sulfatide Analysis Service
Agilent 6495C Triple Quadrupole Mass Spectrometer
- Ultra-sensitive MRM mode with femtogram-level detection
- High dynamic range (up to 6 orders of magnitude) for quantifying both major and minor sulfatide species
- Excellent ion ratio reproducibility and low matrix effect interference
Agilent 1290 Infinity II UHPLC System
- High-resolution reverse-phase separation for lipid classes
- Retention time reproducibility with RSD < 0.5%
- Supports rapid cycle times while maintaining peak integrity
Advanced Software Platforms
- MassHunter Quantitative Analysis (Agilent) for MRM data integration
- LipidSearch™ / Progenesis QI™ for identification and lipid classification
- In-house QC and statistical pipelines for PCA, clustering, and pathway mapping

Agilent 1290 UHPLC System (Figure from Agilent)

Agilent 6495C Triple Quadrupole (Figure from Agilent)
Sample Requirements for Sulfatide Analysis Service
| Sample Type | Minimum Amount Required | Storage Condition | Notes |
|---|---|---|---|
| Plasma / Serum | ≥ 200 µL | -80°C | Avoid hemolysis; use EDTA or heparin tubes |
| Tissue (e.g., brain, liver) | ≥ 50 mg | Snap-frozen, -80°C | Remove blood contamination if possible |
| Cultured Cells | ≥ 2 × 10⁶ cells | Cell pellet, -80°C | Wash with PBS before freezing |
| CSF (Cerebrospinal Fluid) | ≥ 300 µL | -80°C | Preferably collected in polypropylene tubes |
| Urine | ≥ 500 µL | -80°C | Collect midstream sample; centrifuge before freezing |
| Isolated Lipid Extracts | ≥ 50 µg total lipid | Dry film or in organic solvent, -20°C or lower | Indicate solvent composition upon submission |
| Feces (Optional) | ≥ 100 mg | -80°C | Use sterile containers; avoid preservatives |
Applications of Sulfatide Assay Service
Lipidomics-Based Functional Studies
Investigate the role of sulfatides in membrane fluidity, vesicle trafficking, and protein-lipid interactions in various biological systems.
Developmental Biology Research
Profile sulfatide dynamics during embryonic development, neural differentiation, and tissue maturation in model organisms.
Comparative Species Studies
Analyze sulfatide composition across species to explore evolutionary conservation and divergence in sphingolipid metabolism.
Environmental and Nutritional Influence Assessment
Evaluate how external factors such as diet, stress, or toxins affect sulfatide biosynthesis and degradation pathways.
Biomolecular Pathway Mapping
Integrate sulfatide profiles with transcriptomics and proteomics data to elucidate their role in glycosphingolipid and lipid signaling pathways.
Quality Control in Bioprocessing and Lipid Manufacturing
Monitor sulfatide levels as stability markers or process indicators in lipid-based product development and cell culture systems.
Demo

Mass spectral comparison of sulfatide molecular species present in mouse brain cortical lipid extracts acquired by either MALDI-MS or ESI-MS. (Cheng, Hua, et al., 2010).
FAQ of Sulfatide Analysis Service
Can I use this data to correlate sulfatides with other omics datasets?
Yes. Our output is fully compatible with multi-omics integration. We can align sulfatide quantification data with your transcriptomics, proteomics, or metabolomics datasets to enable systems-level analysis.
How stable are sulfatides in biological samples? Will freeze-thaw cycles affect them?
Sulfatides are relatively stable under -80°C storage, but multiple freeze-thaw cycles can degrade them or affect quantification. We recommend aliquoting your samples before shipping to avoid degradation.
Can you differentiate isobaric sulfatide species or regioisomers?
Yes. Using MS/MS fragmentation and retention time separation on our UHPLC-QqQ platform, we can distinguish structural isomers and isobars, especially hydroxylated vs. non-hydroxylated species.
Is there a recommended normalization strategy for comparing sulfatides across tissue types?
We offer multiple normalization options (e.g., total lipid content, protein concentration, or cell number) depending on the matrix and study design. We also provide guidance based on your experimental context.
How do you ensure reproducibility across large sample cohorts?
We use internal quality control samples, pooled biological replicates, and instrument QC calibration every 10–15 runs. CVs are tightly controlled to ensure consistency across large datasets.
Can I submit previously extracted lipid samples? Will that affect the analysis?
Yes, you can submit lipid extracts. Please ensure they are stored in organic solvents under inert conditions and submit full extraction details. The analysis quality depends on lipid stability and sample preparation.
What if I need to analyze very rare or custom sulfatide species?
We can accommodate requests for rare sulfatides by adjusting MRM transitions or including custom standards, if available. Custom method development may extend project timelines slightly.
Is it possible to validate sulfatide changes in a pilot study before full-scale analysis?
Yes. We offer pilot-scale assays for method feasibility, early data insights, and budget-conscious decision-making prior to large-scale sample submission.
Learn about other Q&A.
Sulfatide Analysis Service Case Study

Publications
Here are some publications in Metabolomics research from our clients:

- Loss of G0/G1 switch gene 2 (G0S2) promotes disease progression and drug resistance in chronic myeloid leukaemia (CML) by disrupting glycerophospholipid metabolism. 2022. https://doi.org/10.1002/ctm2.1146
- Multi‐omics identify xanthine as a pro‐survival metabolite for nematodes with mitochondrial dysfunction. 2019. https://doi.org/10.15252/embj.201899558
- Experimental microbial dysbiosis does not promote disease progression in SIV-infected macaques. 2018. https://doi.org/10.1038/s41591-018-0132-5
- Glucosylceramide is essential for Heartland and Dabie bandavirus glycoprotein-induced membrane fusion. 2023. https://doi.org/10.1371/journal.ppat.1011232
- Laboratory evaluation of larvicidal and oviposition deterrent properties of edible plant oils for potential management of Aedes aegypti (Diptera: Culicidae) in drinking water containers. 2019. https://doi.org/10.1093/jme/tjz021
Reference
- Cheng, Hua, et al. "Selective desorption/ionization of sulfatides by MALDI-MS facilitated using 9-aminoacridine as matrix." Journal of lipid research 51.6 (2010): 1599-1609. https://doi.org/10.1194/jlr.D004077







