Protein Hydroxylation Analysis Service
Creative Proteomics offers protein hydroxylation analysis services using high-resolution mass spectrometry to identify and map specific hydroxylation sites on proteins. The service includes sample preparation, targeted enrichment of hydroxylated peptides, and comprehensive data analysis to provide detailed insights into protein modifications. This approach supports research in areas like protein structure, function, and regulation, with no need for complex labeling, ensuring high sensitivity and accuracy.
Submit Your Request Now
×- Define
- What We Provide
- Technology Platform
- Workflow
- Advantages
- Sample Requirements
- Demo
- FAQs
- Case Study
- Publications
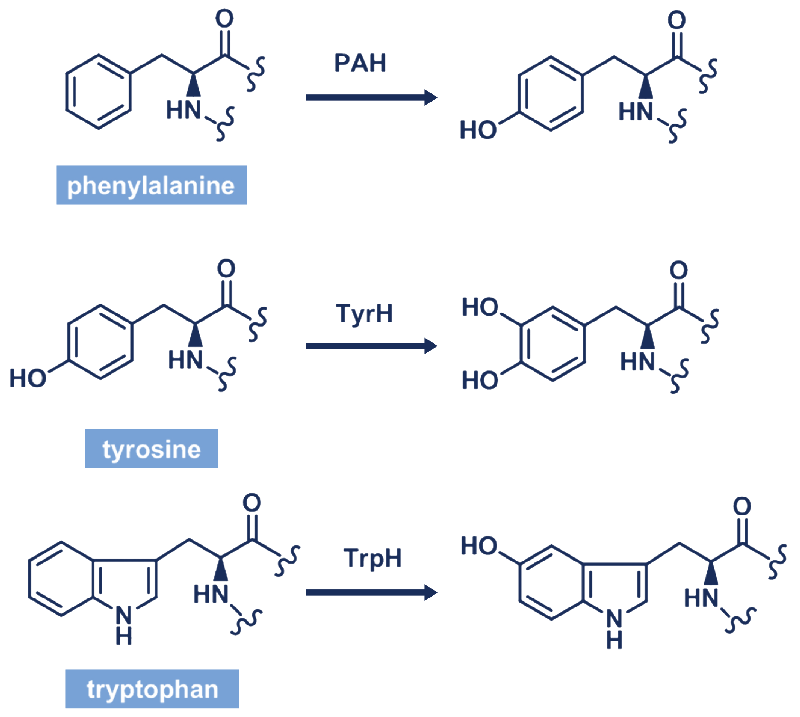
What Is Protein Hydroxylation?
Protein hydroxylation is an essential post-translational modification that involves the introduction of a hydroxyl group (-OH) into amino acid residues within proteins. This modification, catalyzed by dioxygen-dependent enzymes, plays a critical role in regulating protein structure, function, and stability. Hydroxylation also enhances the hydrophilicity of proteins, making them more soluble in aqueous environments and aiding in their clearance by the kidneys and liver. It typically occurs on amino acids such as proline, lysine, and phenylalanine, influencing their structural conformation and enabling specific molecular interactions. For instance, the hydroxylation of proline is crucial for maintaining the triple-helical structure of collagen.
Functions of Protein Hydroxylation
Hydroxylation plays a very important role in the structure and function of organic molecules and biomolecules. It converts hydrophobic molecules into hydrophilic molecules, thus enhancing the overall solubility. In addition, hydroxylated molecules preserve better cleavage by the kidneys and liver, so they tend to excrete more easily than non-hydroxylated molecules.
To elaborate further, hydroxylation converts phenylalanine (Phe) residues to tyrosine (Tyr) residues, thus playing an important role in controlling the excess of Phe residues in the organism. Hydroxylation plays an important role in the conversion of Try to I-DOPA, which is important for the biosynthesis of dopamine. Neurotransmitters such as epinephrine are catecholamine hormones derived from dopamine. Therefore, hydroxylation is essential for the production of neurotransmitters. Nevertheless, proline hydroxylation plays an important role in the homeostasis of protein structure.
Protein Hydroxylation Mass Spectrometry Detection Service
Hydroxylation adds an oxygen atom with a mass of 16 Da, and this increase in molecular weight can be easily detected by high-resolution mass spectrometry. Relying on the professional HPLC-MS/MS mass spectrometry platform, Creative Proteomics can efficiently and accurately identify protein hydroxylation sites in eukaryotic and prokaryotic samples.
Key Features of Our Protein Hydroxylation Analysis Service:
High Sensitivity and Precision Our service utilizes high-resolution mass spectrometry to detect even the most subtle hydroxylation modifications. This allows for the identification of low-abundance hydroxylated peptides, ensuring comprehensive results even from small or complex samples.
Comprehensive Hydroxylation Site Mapping We identify specific hydroxylation sites on proteins, providing detailed mapping of these modifications. This can include modifications on amino acids like proline, lysine, and phenylalanine, which are critical for protein structure and function.
Advanced Sample Preparation We offer two approaches for sample preparation:
- In-solution digestion: Ideal for soluble proteins.
- In-gel digestion: Suitable for proteins separated via gel electrophoresis.
This flexibility allows us to handle various sample types and ensure the best strategy for optimal results.
Targeted Enrichment for Hydroxylated Peptides Our service includes targeted enrichment techniques to isolate hydroxylated peptides, increasing the detection sensitivity for specific modifications and improving the overall coverage of hydroxylation sites.
Comprehensive Data Analysis We provide thorough data analysis, using specialized software to interpret mass spectrometry results. Our expert team will deliver detailed reports that include hydroxylation site identification, peptide sequences, and modifications, along with associated biological implications.
No Need for Complex Labeling Unlike methods requiring isotopic labeling, our mass spectrometry-based approach does not rely on complex pre-processing, making the service simple and cost-effective while maintaining high accuracy.
Protein Hydroxylation Analysis Technology Platform
High-Performance Liquid Chromatography (HPLC)
Agilent 1260 Infinity II HPLC System
This system is used for the separation of peptides before mass spectrometry analysis. It provides high resolution and reproducibility, ensuring effective peptide separation for optimal identification of hydroxylated peptides.
Mass Spectrometry (MS/MS)
Thermo Scientific Orbitrap Fusion Lumos Tribrid Mass Spectrometer
The Orbitrap Fusion Lumos is a cutting-edge mass spectrometer offering high resolution and sensitivity for complex proteomics analysis. It provides accurate mass measurements, essential for identifying hydroxylation sites with high specificity.
Key Features:
- High-resolution MS for precise peptide mass determination
- Ion trap for fragmentation (MS/MS) to identify peptide sequences and modifications
- Advanced data analysis capabilities for post-translational modification identification
AB SCIEX TripleTOF 6600
The TripleTOF 6600 combines high sensitivity with high resolution, delivering accurate, quantitative analysis of modified proteins, including those with hydroxylation.
Key Features:
- Accurate mass and time (AMT) tagging for modification identification
- Enhanced sensitivity for low-abundance peptides
- Deep coverage of post-translational modifications
Data Analysis Software
Software: Proteome Discoverer (Thermo Fisher) / MaxQuant
These advanced bioinformatics tools help process raw mass spectrometry data, identify hydroxylation sites, and quantify the modification levels. They use sophisticated algorithms to assign hydroxylation modifications to specific peptides, offering accurate and detailed analysis.
Software: PEAKS Studio
Used for peptide identification and de novo sequencing. It helps to detect and analyze post-translational modifications, including hydroxylation, by comparing experimental data with theoretical peptide sequences.



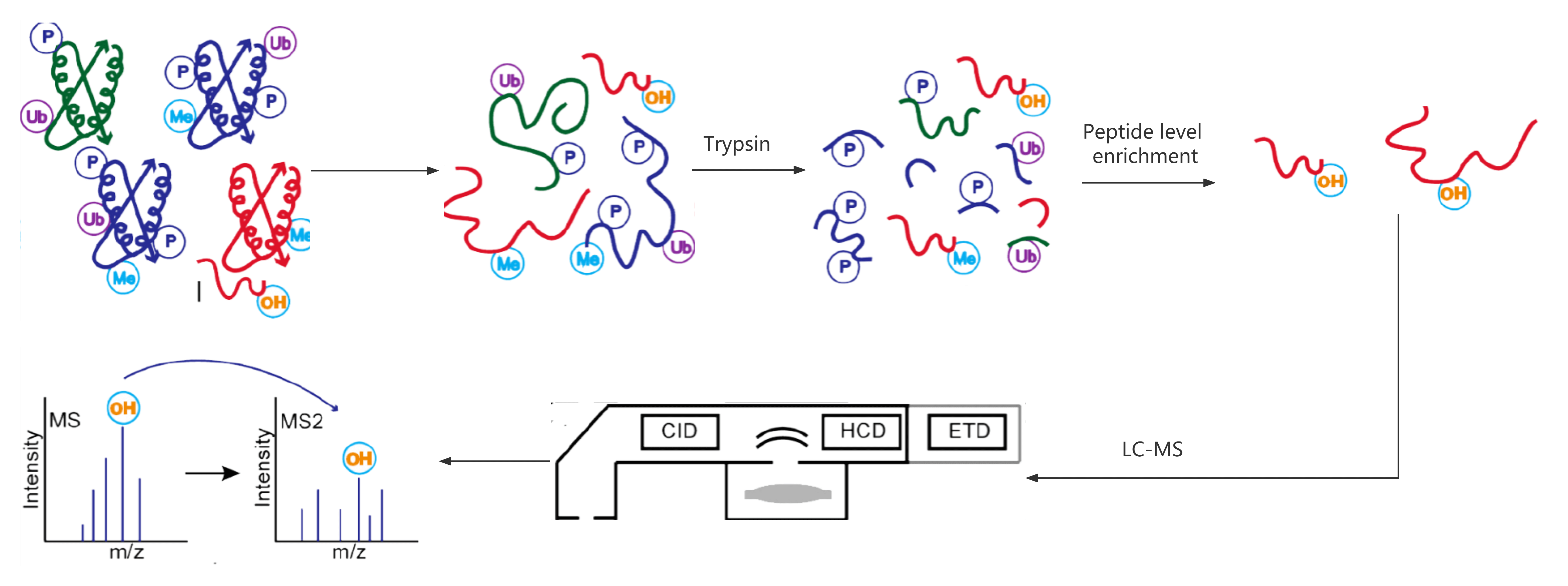
Workflow of Protein Hydroxylation Analysis Service
a) Sample Preparation
Proteins are digested either in-solution or in-gel to break down into peptides suitable for analysis.
b) Peptide Enrichment
Hydroxylated peptides are selectively enriched, improving the detection of hydroxylation modifications.
c) HPLC Separation and Mass Spectrometry
Peptides are separated using high-performance liquid chromatography (HPLC), followed by mass spectrometry analysis (MS/MS) to identify hydroxylation sites.
d) Data Interpretation
The data is analyzed using advanced bioinformatics tools to pinpoint specific hydroxylation sites and provide insight into their functional roles.
Advantages of Protein Hydroxylation Analysis
- Wide range of applications: characterization of hydroxylation sites for various proteins; identification of specific modification sites and specifically modified peptides
- Simple and convenient operation: no radioisotope labeling or other complex pre-processing require
- High site coverage (optional): enrichment of hydroxylated target proteins for increased detection coverage
Sample Requirements for Protein Hydroxylation Analysis
| Sample Type | Requirement |
|---|---|
| Tissue Samples | |
| - Plant Tissue | > 400 mg |
| - Blood (with EDTA for plasma) | ≥ 2 mL |
| - Serum | 2 mL |
| - Urine | 10 mL |
| - Animal Tissue | ≥ 2 g |
| - Cell Samples | 1 x 10^8 cells |
| - Yeast, Microorganisms, Others | Dry weight of 400 mg |
| Protein Sample | Total protein of 2-5 mg (tissue or cell lysate can be used) |
Demo
Bar Chart of Total Protein Identification
2D PCA Plot of Sample Grouping
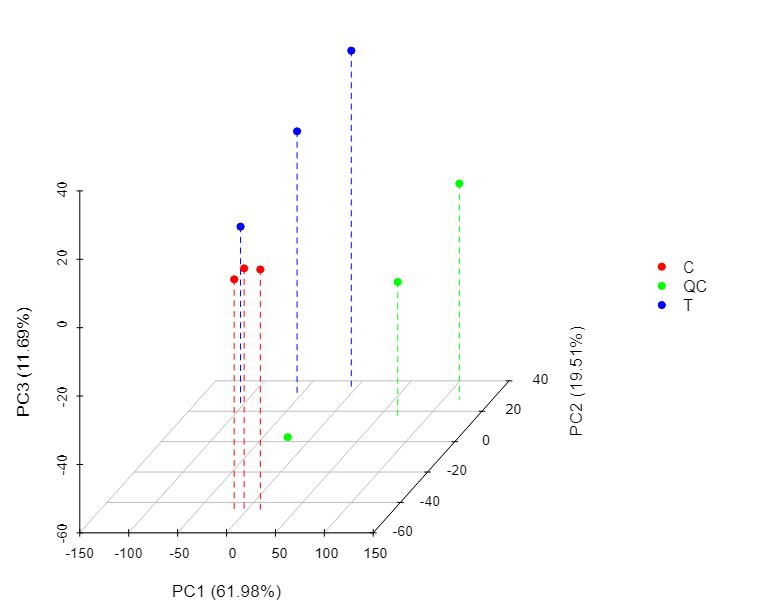
3D PCA Plot of Sample Grouping
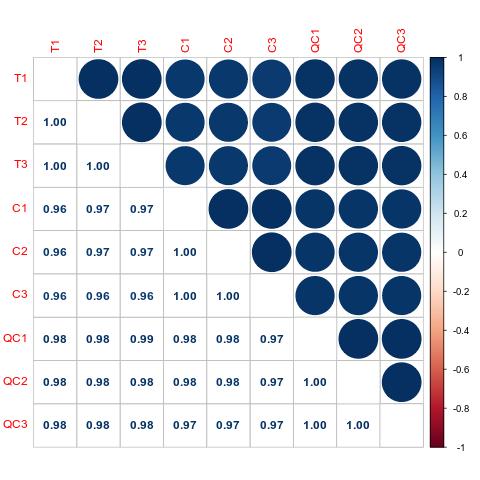
Pearson Correlation Analysis
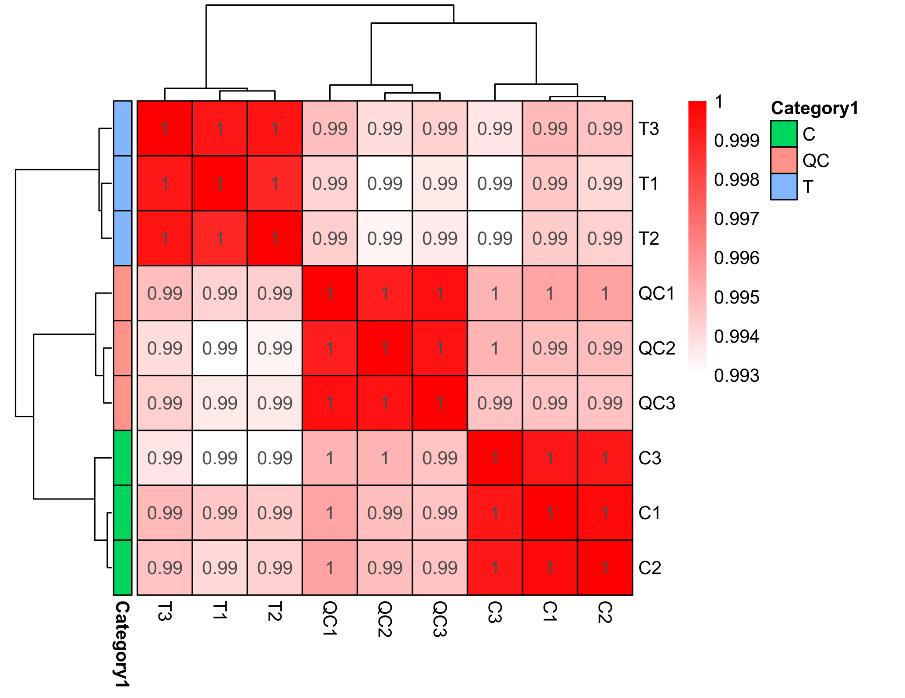
Sample Hierarchical Clustering
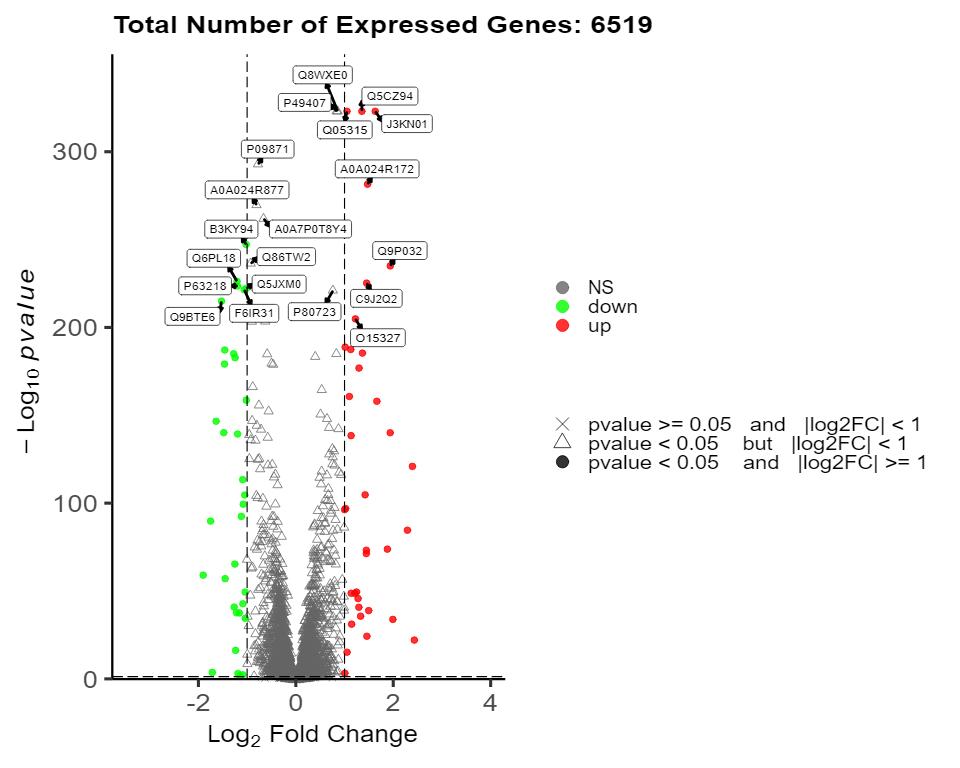
Volcano Plot of Differential Proteins
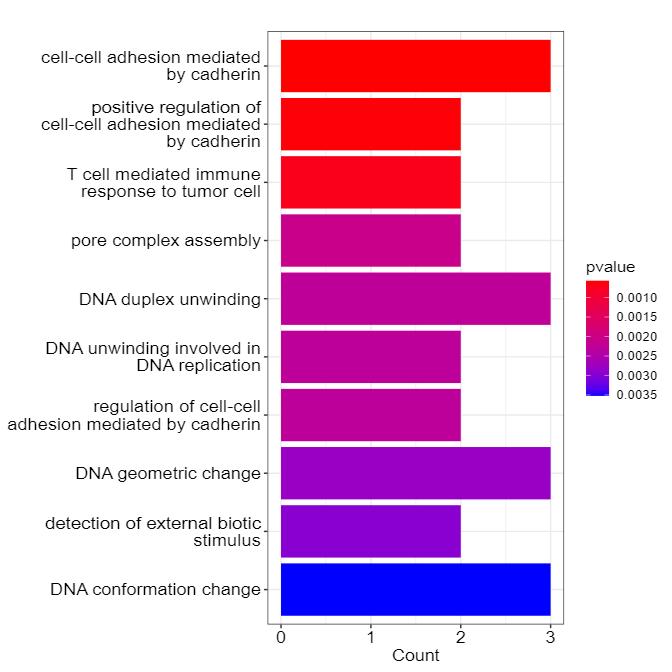
Bar Chart of GO Enrichment for Candidate Proteins
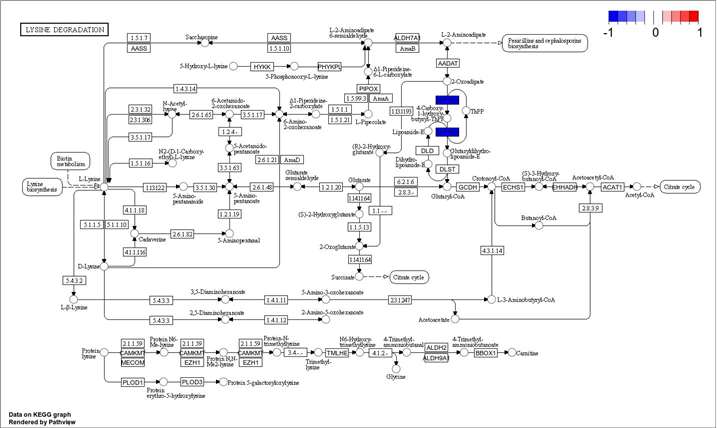
KEGG Pathway Enrichment of Candidate Proteins
FAQs
What types of proteins can be analyzed for hydroxylation?
Our Protein Hydroxylation Analysis Service can be applied to a wide variety of proteins, including soluble and membrane-bound proteins, as well as complex protein mixtures. Hydroxylation typically occurs on residues such as proline, lysine, and phenylalanine, but the modification is not limited to these amino acids. We can analyze proteins from various organisms, including eukaryotic, prokaryotic, and even microbial species, as long as the proteins are adequately extracted and prepared.
How do you ensure the detection of low-abundance hydroxylated peptides?
To detect low-abundance hydroxylated peptides, we employ targeted peptide enrichment techniques. This step increases the relative abundance of hydroxylated peptides before the mass spectrometry analysis. Additionally, we utilize high-resolution mass spectrometry platforms like the Thermo Scientific Orbitrap Fusion Lumos and AB SCIEX TripleTOF 6600, which offer exceptional sensitivity, even for peptides at low concentrations. These instruments can differentiate hydroxylation from other potential modifications by accurately measuring mass shifts as small as 16 Da, ensuring detection even in complex samples.
Can I analyze hydroxylation sites on a specific protein of interest in a complex sample?
Yes, our service includes targeted enrichment methods that allow us to isolate and concentrate hydroxylated peptides from complex mixtures. By selectively enriching for hydroxylated peptides, we can increase the likelihood of detecting specific modifications on a protein of interest, even when the protein is present at low concentrations within a complex sample. This targeted approach is particularly useful for identifying hydroxylation on proteins that may be otherwise challenging to analyze due to low abundance or interference from other proteins.
How do you differentiate hydroxylation from other modifications like phosphorylation or methylation?
Hydroxylation is characterized by the addition of a single oxygen atom (16 Da) to the peptide, which can be distinguished from other post-translational modifications (PTMs) by mass spectrometry. For instance:
- Phosphorylation adds a phosphate group (80 Da), causing a different mass shift.
- Methylation typically adds a methyl group (14 Da) to the peptide.
Our mass spectrometry platform offers the sensitivity required to detect subtle mass shifts and differentiate between these modifications. Moreover, advanced data analysis tools, such as Proteome Discoverer or MaxQuant, are used to compare experimental data with theoretical modifications, ensuring precise identification of hydroxylation and other PTMs.
Learn about other Q&A.
Case Study

LC-MS/MS Identification of the O-Glycosylation and Hydroxylation of Amino Acid Residues of Collagen α-1 (II) chain from Bovine Cartilage
Journal: Journal of Proteome Research
Published: 2013
- Background
- Materials & Methods
- Results
- Reference
Collagen is a critical structural protein found in the extracellular matrix of vertebrates, with type II collagen (CO2A1) being the primary component of cartilage and vitreous humor. The post-translational modifications (PTMs) of collagen, including hydroxylation and O-glycosylation, play significant roles in collagen's structural stability, fibril formation, and biological functions. Hydroxylation of proline and lysine residues (specifically hydroxyproline and hydroxylysine) is essential for stabilizing the collagen triple helix, while O-glycosylation of hydroxylysine residues influences collagen cross-linking and fibril assembly. These modifications are critical for proper collagen function and can vary among tissue types and collagen subtypes.
In this study, LC-MS/MS was employed to identify and quantify the O-glycosylation and hydroxylation of the amino acid residues of bovine type II collagen α-1 chain. Specifically, 23 O-glycosylation sites were identified, along with numerous hydroxyproline (HyP) residues in the collagen structure. The study also highlighted the occurrence of deamidation of asparagine residues in collagen, which results in the conversion to aspartic acid (Asp) and isoaspartic acid (isoAsp). Through the use of multiple fragmentation techniques such as CID, ETD, and HCD, the study provided a comprehensive analysis of the glycosylation and hydroxylation patterns, revealing important insights into collagen's post-translational modifications and their biological implications.
Materials
- Bovine collagen α-1 (II) chain (CO2A1): Pepsinized, immunization grade, from Chondrex.
- Chemicals: Ammonium bicarbonate, sodium phosphate, MS-grade formic acid from Sigma-Aldrich; HPLC-grade solvents (methanol, isopropanol, acetonitrile) from Fisher Scientific; water from Mallinckrodt.
- Enzymes: Trypsin Gold (Promega), Endoproteinase GluC (New England Biolabs).
Enzymatic Digestion
Sample preparation: 20 µg of CO2A1 was digested using GluC (1:20 enzyme/substrate ratio) in PBS or Trypsin (1:50 enzyme/substrate ratio) in ammonium bicarbonate buffer. Samples were denatured at 65°C for 2 hours and digested at 37.5°C for 16 hours. Digestion was quenched with formic acid.
LC-MS/MS Analysis
- Chromatography: Peptides were separated using a Dionex nano-LC system with a PepMap 100 C18 capillary column, at a flow rate of 350 nl/min. Gradient: 0-50 min with increasing solvent B (ACN + 0.1% formic acid).
- Mass Spectrometry: LTQ Orbitrap Velos equipped with nano-ESI source. MS scan range 380–2000 m/z, followed by CID and HCD MS/MS for peptide fragmentation. ETD was also used for glycopeptide analysis. Three scans were performed: full MS scan, CID MS/MS, and HCD MS/MS.
Data Processing and Quantification
- Identification: Peptide identification was done using Mascot and Scaffold software against the SwissProt database, with variable modifications (hydroxylation, glycosylation).
- Quantification: Peak heights were determined using Xcalibur Qual Browser. Internal standards (specific peptides) were used for normalization to account for ESI spray variation.
Enzymatic Digestion:
- GluC and trypsin were used for CO2A1 digestion, achieving 90% sequence coverage.
- GluC cleaves at glutamic acid and aspartic acid, while trypsin cleaves at lysine and arginine.
Identification of PTMs:
- 23 out of 24 glycosylation sites were identified in CO2A1.
- Found 104 hydroxylated prolines (HyP) and 14 hydroxylated lysines (HyK).
- Identified novel modifications, such as a HyP at Gly-HyP-Gln (P220), not previously reported.
Comparison with Human CO2A1:
- 98% homology between bovine and human CO2A1, but differences in PTMs (e.g., K773 was unmodified in bovine, modified in human).
Micro- and Macroheterogeneity of Glycosylation:
- Multiple glycoforms were observed at K299/K308 and K452/K464, showing variation in glycan composition.
- Macroheterogeneity observed with different glycosylation patterns across protein isoforms.
Semi-Quantification of Modifications:
- Semi-quantitative analysis showed variations in glycosylation levels, with K773 100% unmodified and K884 mainly glycosylated.
- Glycosylation correlated with hydroxylation levels and steric hindrance.
Biological Implications:
- PTMs likely influence fibrillogenesis, cross-linking, and matrix mineralization.
- Insights into these modifications could aid understanding of collagen-related diseases like rheumatoid arthritis.
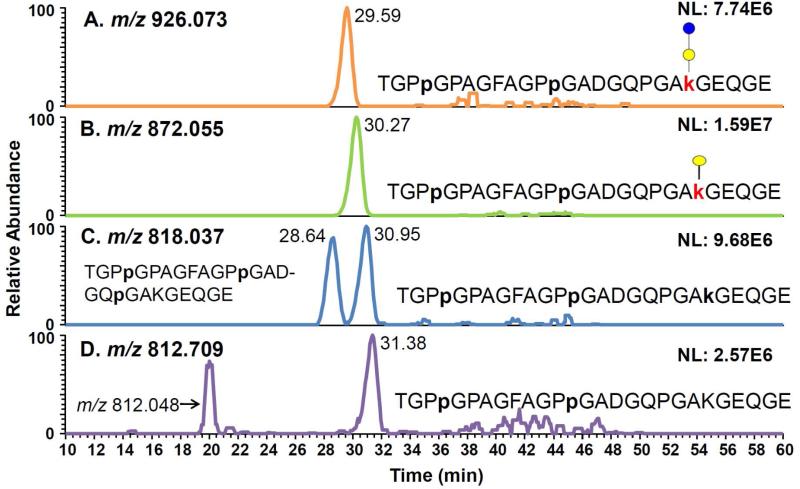 EICs of TGPpGPAGFAGPpGADGQPGAkGEQGE peptide with modifications of Glc-Gal(A), Gal(B), and HyK(B) and unmodified K(D).
EICs of TGPpGPAGFAGPpGADGQPGAkGEQGE peptide with modifications of Glc-Gal(A), Gal(B), and HyK(B) and unmodified K(D).
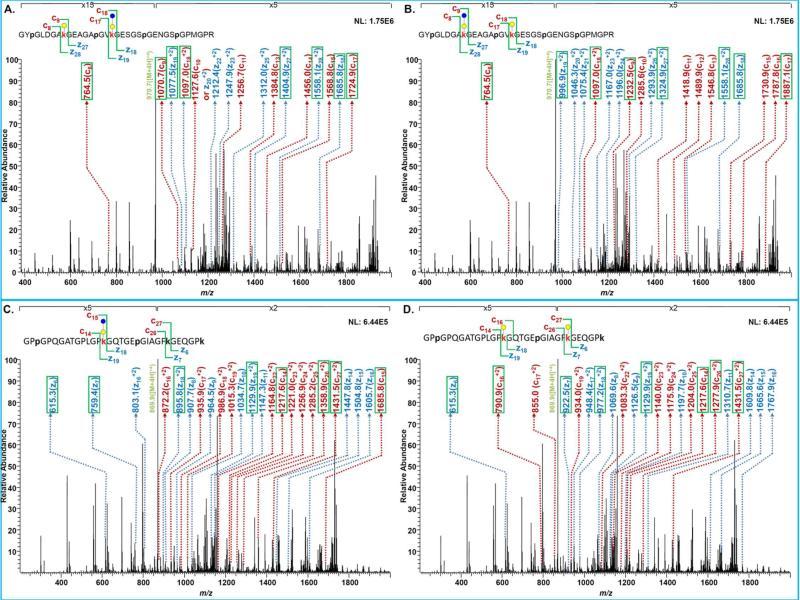 CID tandem MS of the peptide TGPPGPAGFAGPPGADGQPGAKGEQGE modified with different moieties on K848. (A) Peptide modified with Glc-Gal moiety at K848. (B) Peptide modified with Gal moiety at K848. (C) Peptide modified with HyP but not HyK at K848. (D) Peptide modified with both HyP and HyK at K848. (E) Peptide with no modification at K848.
CID tandem MS of the peptide TGPPGPAGFAGPPGADGQPGAKGEQGE modified with different moieties on K848. (A) Peptide modified with Glc-Gal moiety at K848. (B) Peptide modified with Gal moiety at K848. (C) Peptide modified with HyP but not HyK at K848. (D) Peptide modified with both HyP and HyK at K848. (E) Peptide with no modification at K848.
Reference
- Song, Ehwang, and Yehia Mechref. "LC–MS/MS identification of the O-glycosylation and hydroxylation of amino acid residues of collagen α-1 (II) chain from bovine cartilage." Journal of proteome research 12.8 (2013): 3599-3609.
Publications
Here are some publications in Proteomics research from our clients:

- Epithelial WNT2B and Desert Hedgehog are necessary for human colonoid regeneration after bacterial cytotoxin injury. 2018. https://doi.org/10.1101/434639
- Metasecretome and biochemical analysis of consortium PM-06 during the degradation of nixtamalized maize pericarp. 2023. https://doi.org/10.1016/j.bcab.2023.102634
- Calcium homeostasis and stable fatty acid composition underpin heatwave tolerance of the keystone polychaete Hediste diversicolor. 2021. https://doi.org/10.1016/j.envres.2021.110885
- Mechanistic and biomarker studies to demonstrate immune tolerance in multiple sclerosis. 2022. https://doi.org/10.3389/fimmu.2021.787498
- APOB100 transgenic mice exemplify how the systemic circulation content may affect the retina without altering retinal cholesterol input. 2024. https://doi.org/10.1167/iovs.61.13.19





