Histone Post-Translational Modification Analysis Service
Histone proteins play a critical role in packaging DNA and controlling gene activity. Despite being highly conserved, their N-terminal tails are hotspots for a wide range of post-translational modifications (PTMs)—including methylation, acetylation, phosphorylation, ubiquitination, propionylation, ADP-ribosylation and other modifications. These chemical tags act as signals that reshape chromatin structure and influence how genes are turned on or off.
At Creative Proteomics, our histone PTMs analysis service is designed to help researchers decode these modifications with precision. Whether you're investigating tumor progression or stem cell differentiation, our advanced proteomics workflows provide actionable insights into chromatin regulation.
Submit Your Request Now
×- Experimental procedures
- Mass Spectrometry Parameters
- Details of histone PTMs
- Mass Spectrogram
- Technique Comparison
- Service Details
- Delivery&Demo
- FAQ
- Case Study
- Publication
Why Histone Modifications Matter
Histone modifications don't change the underlying DNA sequence. Instead, they alter the physical and chemical properties of histone proteins, affecting how tightly DNA is wound around them. These changes:
- Regulate key processes like DNA replication, repair, and transcription
- Recruit transcription factors or chromatin-binding proteins
- Influence cell fate decisions and disease outcomes, particularly in cancer
Because of their ability to reshape gene expression without altering DNA, histone PTMs are now considered a core component of the epigenetic code. Abnormal patterns in these modifications are closely linked to tumorigenesis, making them valuable biomarkers and therapeutic targets.
Comparative Overview of Histone Modification Detection Techniques
| Technique | Principle | Advantages | Limitations | Optimal Applications |
|---|---|---|---|---|
| ChIP-on-chip | Combines chromatin immunoprecipitation (ChIP) with DNA microarray analysis. |
|
|
|
| ChIP–SAGE | Integrates ChIP with Serial Analysis of Gene Expression for tag-based profiling. |
|
|
|
| ChIP–Seq | Combines ChIP with high-throughput sequencing for precise mapping. |
|
|
|
| Native ChIP (NChIP) | Uses native chromatin without cross-linking for immunoprecipitation. |
|
|
|
| Cross-linking ChIP (XChIP) | Involves cross-linking proteins to DNA before immunoprecipitation. |
|
|
|
| ELISA-based Detection | Utilizes antibodies to detect specific histone modifications in a plate-based assay. |
|
|
|
| Mass Spectrometry (MS) | Detects and quantifies histone modifications based on mass-to-charge ratios. |
|
|
|
The Analytical Challenge—And Our Solution
Histone PTMs are complex: they exist in multiple forms, vary between cell types, and shift rapidly in response to stimuli. Compounding this, histone-binding proteins are often low in abundance and weakly associated with chromatin, making detection difficult.
To address this, Creative Proteomics has developed a sensitive and systematic solution for histone PTM profiling. Our platform integrates:
Nano-LC coupled with high-resolution mass spectrometry (MS)
- Advanced enrichment and labeling techniques for modified peptides
- Proprietary sample preparation methods to preserve labile modifications
This workflow has been validated in diverse biological systems, from cancer cells to stem cell lines.
Main benefits of MS include:
- Fast analysis of many PTMs in one test.
- High resolution over 60,000 for exact modification detection.
- Measuring MS signals to find amounts of analytes.
What We Offer
- Identification of histone modification types and their precise locations
- Quantitative comparison across different cell states or treatment conditions
- Bioinformatics interpretation of functional pathways affected by histone marks
All we need is your sample and project goal—our expert team will take care of the rest.
What PTMs We Analyse
Creative Poteomics quantifies key modifications, including but not limitied:
- Acetylation: Associated with gene activation
- Methylation: Can activate or repress depending on site
- Phosphorylation: Linked to stress response and cell cycle
- Ubiquitination: Influences DNA repair and histone degradation
- ADP-ribosylation, and other modifications.
From Sample to Insight: Our Complete PTM Workflow
Step-by-Step Overview
Our service begins with careful sample preparation to protect histone integrity. Then, we:
- Isolate & purify histones
- Use multi-enzyme digestion to maximise PTM site coverage
- Run LC-MS/MS for ultra-sensitive detection
- Apply advanced bioinformatics for data interpretation
Workflow for Histone Isolation and Purification
Precision Preparation for Reliable PTM Analysis
Accurate analysis of histone post-translational modifications (PTMs) starts with one essential step: isolating pure histone proteins. Unlike conventional proteomic workflows, which can analyse whole-cell lysates, histone PTM studies demand extra precision. These proteins reside deep within the nucleus, tightly bound to DNA, making extraction challenging.
At Creative Proteomics, we've developed a specialised histone isolation platform to meet this challenge. Drawing on validated literature methods and our in-house expertise, we ensure each sample is prepared for maximum sensitivity in downstream mass spectrometry analysis.
Our Histone Extraction Protocol at a Glance
To ensure data accuracy and reproducibility, our workflow follows a robust multi-step protocol:
Step 1: Nuclear Isolation
Cells are lysed and nuclei isolated via differential centrifugation.
Step 2: Histone Extraction
Nuclear membranes are disrupted to release chromatin. A high-salt buffer separates histones from DNA and other nuclear components.
Step 3: Fractionation & Purification
Using high-performance liquid chromatography (HPLC), histones are fractionated into H1, H2A, H2B, H3, and H4 variants.
This step-by-step purification ensures that only clean histone samples—free from DNA, enzymes, and debris—proceed to mass spectrometry.
Optimising for Maximum PTM Coverage
To achieve full-spectrum analysis of histone PTMs, sample preparation must be tailored for modification preservation and peptide coverage. Here's how we do it:
Multi-Enzyme Digestion Strategy
Instead of relying on a single protease, we apply two to three different enzymes to digest histones. This approach increases sequence coverage and ensures detection of short, heavily modified peptides.
Ionisation Efficiency Maximised
By optimising peptide length and charge, we reduce the risk of low ionisation—a common issue in PTM-rich regions.
PTM Retention Ensured
Our protocols are specifically designed to retain labile modifications, such as phosphorylation or ADP-ribosylation, that often go undetected with standard workflows.
Built for High-Confidence MS Analysis
Our refined workflow ensures that histone samples entering the mass spectrometer are both pure and modification-rich—enabling confident, reproducible quantification of histone PTMs.
Whether you're investigating chromatin regulation in cancer, stem cells, or developmental biology, our histone purification platform lays the technical foundation for meaningful discoveries.
Technology Platforms
Thermo Q Exactive HF
Orbitrap Fusion™ / Fusion Lumos™ Mass Spectrometers
Nano-LC for ultra-sensitive peptide separation
![]()
Why Partner with a Professional Histone PTM Analysis Service?
| Benefit | What You Get |
|---|---|
| Accuracy & reliability | High-confidence PTM data validated by MS |
| Access to innovation | Tools like Orbitrap MS, ChIP-seq and ChIP-chip |
| Time and cost savings | Streamlined workflows and expert execution |
| Publication-ready results | Clean datasets for fast reporting & review |
Sample Requirement
1. Biological Samples
To ensure optimal histone extraction and modification profiling, please provide the following minimum quantities:
Plant tissues: ≥ 400 mg
Animal tissues: ≥ 2 g
Blood (EDTA anticoagulated plasma): ≥ 2 mL
Serum: 2 mL
Urine: 10 mL
Cell pellets: ≥ 1 × 10⁸ cells
Microorganisms/Yeast (dry weight): ≥ 400 mg
2. Protein Extracts
We also accept pre-extracted protein lysates from cells or tissues:
Total protein: 2–5 mg
Source: Cell or tissue lysates compatible with standard protein extraction methods
Tip: Ensure samples are free from protease inhibitors and detergents that may interfere with MS.
Shipping Instructions
- Ship all samples on sufficient dry ice to preserve protein integrity.
- Include a completed sample submission form with project ID and contact details.
- For international shipments, ensure compliance with biohazard labeling and customs documentation.
Quality Control Measures
Check sample quality before sending them for analysis. Test antibodies on histone peptide arrays to ensure they work well. Regularly check for cross-reactivity and batch differences to avoid mistakes. These steps improve accuracy and prevent costly errors.
Following these rules ensures your samples meet the needs for advanced histone modification analysis. Careful preparation leads to reliable results, helping your research succeed.
What You'll Receive in the Final Report
We deliver a complete data package tailored for publication, downstream pathway analysis, or therapeutic target validation:
Deliverables
Experimental Protocols
Detailed documentation of sample preparation, histone isolation, and digestion workflow.
Mass Spectrometry Parameters
Including LC-MS/MS setup, instrument settings, and peptide ionisation conditions.
Histone PTM Profiles
Identified modification types, precise amino acid sites, modification intensity, and comparative quantification (if applicable).
Annotated Mass Spectra
High-resolution mass spectrograms with peptide fragmentation maps and scoring data for validation.
Demo

Frequently Asked Questions: Histone PTM Analysis Service
What is histone PTM analysis?
Histone PTM analysis identifies and quantifies chemical modifications—such as acetylation, methylation, and phosphorylation—on histone proteins. These modifications regulate gene expression by altering chromatin structure, helping researchers explore cellular development, signalling pathways, and disease mechanisms.
Why choose Creative Poteomics for this service?
We combine advanced mass spectrometry platforms, smart sample processing workflows, and expert bioinformatics interpretation. Our team delivers high-resolution, site-specific results tailored to your experimental questions—whether you're mapping epigenetic changes in cancer or analysing drug-induced chromatin shifts.
What types of samples are accepted?
We accept a range of biological samples, including:
Cell pellets
Tissue lysates
Purified histone proteins
Tip: Keep samples fresh or store them at -80°C. Always label with details like cell type, treatment, and collection date to ensure accurate results.
How long does the analysis take?
Project timelines vary depending on factors such as sample type, analysis depth, and overall complexity. Once we assess your project requirements, we'll provide a tailored estimate. We also offer flexible scheduling options to accommodate urgent research needs.
What deliverables will I receive?
You'll receive a full report package including:
- Sample preparation and MS workflows
- Raw mass spectrometry data
- Identified peptides and modification sites
- Quantitative analysis reports
- Bioinformatics interpretation of biological pathways
These files are publication-ready and suitable for downstream functional analysis.
Can this service support disease research?
Absolutely. Our histone PTM profiling helps identify epigenetic markers linked to cancer, autoimmune disorders, neurological diseases, and more. It also reveals pathways involved in drug resistance and disease progression—making it ideal for translational and preclinical studies.
How should I prepare my samples for shipment?
- Keep samples cold or frozen (-80°C preferred).
- Use cryoprotectants if needed.
- Label all tubes with sample type, treatment condition, and date.
Follow our sample submission guidelines to avoid processing delays or misidentification.
Can I study multiple histone modifications in one analysis?
Yes. Our workflows are optimised to detect multiple PTM types in a single run—giving you a comprehensive view of histone regulation under your experimental conditions.
What makes Creative Poteomics' PTM service unique?
Our platform is built for precision, scalability, and discovery. With:
- State-of-the-art MS instruments
- Proven enrichment and digestion strategies
- In-house data scientists for bioinformatics support
- We empower you with complete, confident datasets to advance your research.
Learn about other Q&A.
Client Case Study: Unveiling SIRT6's Mono-ADP-Ribosylation Activity

DNA stimulates SIRT6 to mono-ADP-ribosylate proteins within histidine repeats. bioRxiv. 2024 https://doi.org/10.1101/2024.07.31.606047
- Background
- Technical Approach
- Results
- Conclusion
Sirtuin 6 (SIRT6) is a nuclear enzyme known for its role in deacetylating histones, thereby influencing gene repression. Beyond this canonical function, SIRT6 also exhibits mono-ADP-ribosylation (mARylation) activity, particularly in response to DNA damage. However, the specific substrates and conditions under which SIRT6 exerts this activity remained unclear. The study aimed to elucidate the mechanisms activating SIRT6's mARylation function and identify its target.
To investigate SIRT6's mARylation activity, the research team employed a combination of biochemical assays and mass spectrometry analyses. Given the complexity of histone modifications and the need for precise identification of PTMs, the team collaborated with Creative Proteomics. Our Histone PTM Analysis Service provided comprehensive support, including:
- Histone Isolation and Enrichment: Utilizing optimized protocols to extract and purify histones from nuclear fractions, ensuring the preservation of PTMs.
- Mass Spectrometry Analysis: Employing high-resolution instruments like the Orbitrap Fusion Lumos, combined with nano-liquid chromatography, to detect and quantify histone modifications with high sensitivity.
- Data Analysis: Applying advanced bioinformatics tools to interpret mass spectrometry data, identify modification sites, and quantify changes in PTMs.
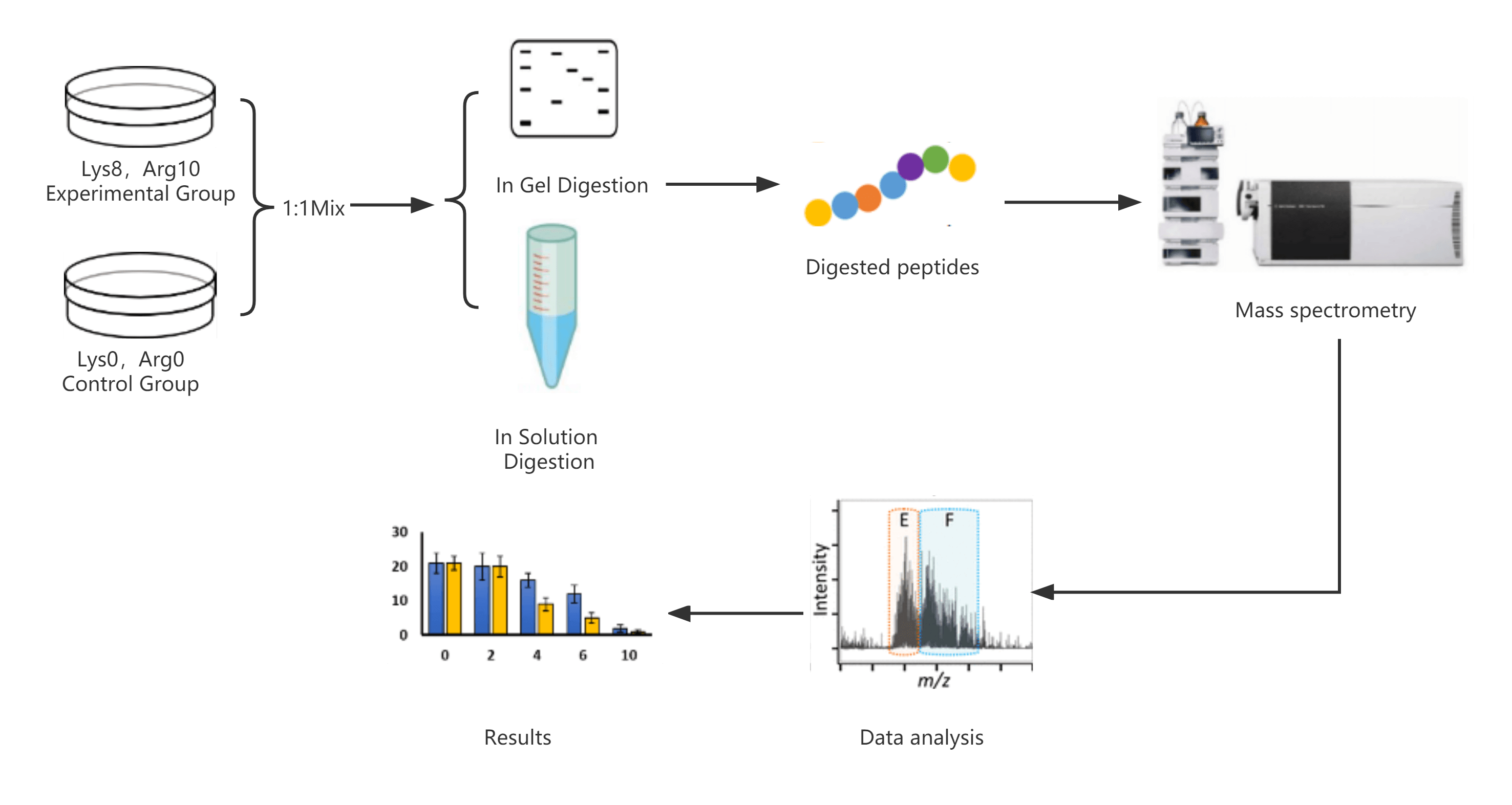
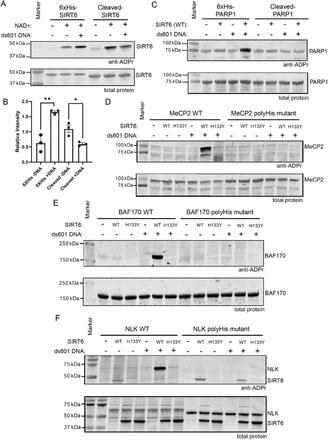 Figure 1: DNA stimulates SIRT6 to mARylate proteins containing polyHis.
Figure 1: DNA stimulates SIRT6 to mARylate proteins containing polyHis.
The collaborative efforts led to several key findings:
- Activation by DNA: SIRT6's mARylation activity was significantly enhanced upon binding to double-stranded DNA ends, suggesting a mechanism where DNA damage signals activate SIRT6.
- Target Motif Identification: Mass spectrometry analyses revealed that SIRT6 preferentially mono-ADP-ribosylates proteins containing polyhistidine (polyHis) repeat tracts. This motif was present in several known SIRT6 substrates and binding partners.
- Distinct from Deacetylase Activity: The mARylation function of SIRT6 was distinct from its deacetylase activity, indicating dual roles in chromatin regulation and DNA repair.
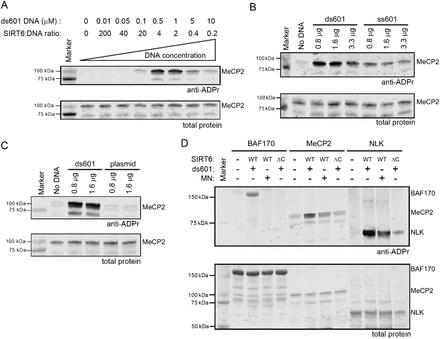 Figure 2: SIRT6 is activated in its mARylation activity by binding to DNA ends
Figure 2: SIRT6 is activated in its mARylation activity by binding to DNA ends
 Figure 3: SIRT6 mARylates Histidine.
Figure 3: SIRT6 mARylates Histidine.
The study provided significant insights into the non-canonical functions of SIRT6, highlighting its role as a histidine-specific mono-ADP-ribosyltransferase activated by DNA damage. These findings contribute to a deeper understanding of chromatin dynamics and the cellular response to genomic stress.
Publication

- Trypanosoma cruzi DNA Polymerase β Is Phosphorylated In Vivo and In Vitro by Protein Kinase C (PKC) and Casein Kinase 2 (CK2).Cells (2022). DOI: https://doi.org/10.3390/cells11223693
- Synergistic Combined-proteomics Guided Mapping strategy identifies mTOR mediated phosphorylation of LARP1 in nutrient responsiveness and dilated cardiomyopathy. bioRxiv (2022). DOI: https://doi.org/10.1101/2022.10.13.512080
- The Interplay between GSK3β and Tau Ser262 Phosphorylation during the Progression of Tau Pathology. International Journal of Molecular Sciences (2022). DOI: https://doi.org/10.3390/ijms231911610
- Exploratory phosphoproteomics profiling of Aedes aegypti Malpighian tubules during blood meal processing reveals dramatic transition in function. PLOS ONE (2022) DOI: https://doi.org/10.1371/journal.pone.0271248












