S-Nitrosation represents a reversible post-translational modification (PTM) characterized by the covalent attachment of nitric oxide (NO) to cysteine (Cys) sulfhydryl groups (-SH), yielding S-nitrosothiol (-SNO) adducts. This redox-sensitive modification serves as a critical regulator of cellular signaling cascades, redox equilibrium, transcriptional control, and metabolic stability. Its physiological significance extends to cardiovascular, neurological, and immunological homeostasis. Dysregulated S-nitrosylation is implicated in diverse pathological states, including neurodegenerative disorders, malignancies, and metabolic diseases such as diabetes.
This review systematically classifies and delineates the molecular mechanisms of protein S-nitrosation, encompassing three principal pathways: (1) direct nitrosative modification, (2) indirect transnitrosation cascades, and (3) non-canonical mechanisms operating under both physiological and pathological regulatory contexts.
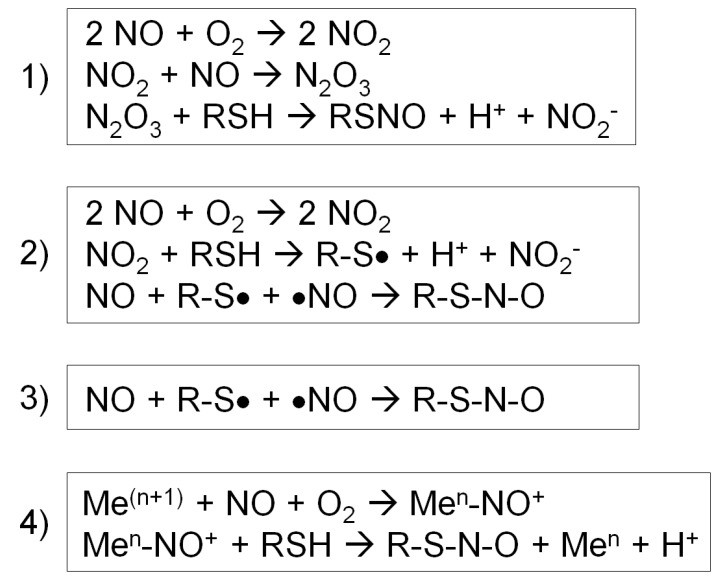 Four different types of S-nitrosylation reactions (Fernando V et al., 2019)
Four different types of S-nitrosylation reactions (Fernando V et al., 2019)
Direct S-Nitrosylation
NO radical pathway
- Mechanism: Nitric oxide radical (NO) directly attacks sulfhydryl group (-SH) of protein cysteine (Cys) to form S- nitrosthiol (SNO).
- Chemical reaction formula:R-SH + NO• → R-SNO
1. Stepwise Copper-Catalyzed Mechanism
Molecular Events:
- Thiol Activation: Acidic microenvironments reduce Cys pKₐ (e.g., adjacent acidic residues), enhancing deprotonation (R-S⁻ formation).
- Metal-Sulfur Coordination: Cu⁺ forms coordination bonds with thiolate anions ([Cu-SR]), amplifying nucleophilicity.
- NO Radical Attack: •NO binds sulfur, generating radical transition state ([Cu²⁺-S(R)NO•]⁻).
- SNO Dissociation: Release of Cu²⁺ stabilizes SNO bond formation (R-SNO).
Iron Catalysis:
- Similar pathway but lower efficiency due to rapid Fe²⁺ oxidation to inactive Fe³⁺.
Non-Catalytic Limitations:
- Requires non-physiological conditions: high NO (>1 μM) and extreme hydrophobicity (e.g., hemoglobin heme pockets).
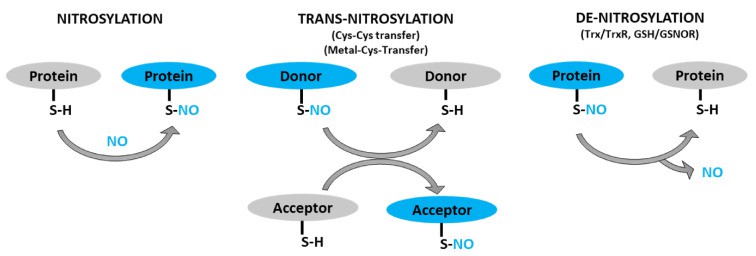 Schematic representation of protein S-nitrosylation, transnitrosylation, and denitrosylation (Sharma V et al., 2021)
Schematic representation of protein S-nitrosylation, transnitrosylation, and denitrosylation (Sharma V et al., 2021)
2. Structural Microenvironment Determinants
Key Regulatory Factors:
- Acidic pH (4.0–6.5): Lowers Cys pKₐ by 2–3 units, promoting deprotonation (e.g., lysosomal Cathepsin B Cys108).
- Hydrophobic Pockets: Exclude water, stabilizing radical intermediates (e.g., hemoglobin β-subunit Cysβ93 heme cavity).
- Electrostatic Networks: Adjacent acidic residues (Asp/Glu) anchor reactive Cys (e.g., PTP1B Cys215).
- Hydrogen Bonding: Donors (e.g., His39) fix SNO conformation (e.g., HSA domain IB Cys34).
3. Physiological Control Systems
- Metal Homeostasis:
- Copper Chaperones: ATOX1/CCS deliver Cu⁺ to nitrosation sites.
- Metallothioneins: Buffer free Cu²⁺ at nanomolar concentrations.
- Spatiotemporal NO Regulation:
- Compartmentalization: eNOS colocalizes with Cu/Zn-SOD1 in caveolae for localized •NO–Cu⁺ coupling.
- Mitochondrial Crosstalk: Superoxide (respiratory chain) → H₂O₂ → NOS activation → •NO bursts.
- Oxygen-Dependent Allostery (Hemoglobin):
- Deoxygenated (T) State: Exposed Cysβ93 binds •NO → SNO formation → vasodilation.
- Oxygenated (R) State: Buried Cysβ93 prevents •NO waste.
4. Pathophysiological Implications
Mitochondrial Complex I (CI) S-Nitrosation:
- Target: Conserved ND3 subunit Cys39.
- Consequences:
- Blocks electron transfer → ATP synthesis collapse.
- Increases O₂•⁻ leakage → mitophagy induction.
- Disease Links:
- Myocardial ischemia-reperfusion injury.
- MitoSNO, a mitochondria-targeted S-nitrosating agent, transiently stabilizes the Cys39 residue within the ND3 subunit in a low-activity conformation through site-specific S-nitrosation, thereby attenuating reactive oxygen species (ROS) generation. This protective mechanism demonstrates particular efficacy in ischemic preconditioning models, underscoring the therapeutic significance of regulated S-nitrosation in myocardial preservation strategies.
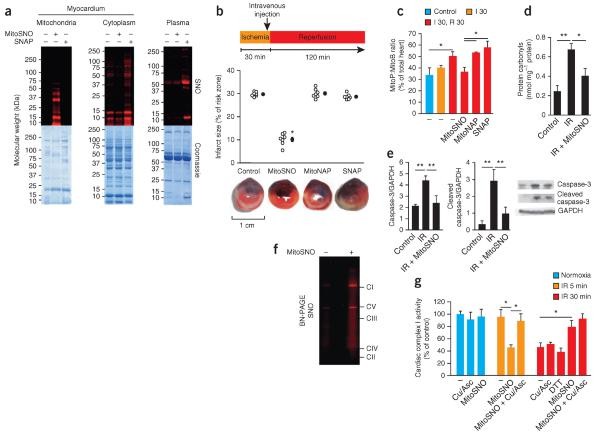 S-nitrosation of mitochondrial proteins is required for S-nitrosation–mediated protection from cardiac ischemia-reperfusion injury (Chouchani ET et al., 2013)
S-nitrosation of mitochondrial proteins is required for S-nitrosation–mediated protection from cardiac ischemia-reperfusion injury (Chouchani ET et al., 2013)
5. Technical Challenges and Therapeutic Strategies
- Detection Limitations:
- SNO lability → false negatives.
- Solutions: SNOTRAP probes (alkyne tags covalently capture SNO).
- Targeted Interventions:
- Copper Chelation: Tetrathiomolybdate (TTM) mitigates ischemia-reperfusion injury.
- NOS Inhibition: L-NAME reduces neuroinflammatory S-nitrosation.
- Reductase Activation: Thioredoxin system reverses pathological SNO.
6. Biological Significance and Future Directions
- Core Functions:
- Millisecond-Scale Precision: Metal–microenvironment synergy regulates oxygen transport (Hb SNO) and metabolic switching (CI nitrosation).
- Dual Roles: Physiological SNO protects vasculature; pathological excess triggers cell death.
- Future Frontiers:
- Cryo-EM structural analysis of SNO reaction intermediates.
- Spatially targeted metal chelators for disease modulation.
Indirect Pathways of S-Nitrosation
Indirect S-nitrosation—mediated by nitrosylating agents (e.g., NO⁺ equivalents) or oxidized reactive nitrogen species—represents a physiologically predominant modification mechanism, characterized by enhanced efficiency and site specificity.
Oxidative Pathway: N₂O₃-Mediated Electrophilic Nitrosation
Reaction Cascade:
- N₂O₃ Formation: Nitric oxide (•NO) reacts with oxygen to generate nitrogen dioxide (•NO₂), which dimerizes to dinitrogen trioxide (N₂O₃).
- Electrophilic Attack: N₂O₃, functioning as a nitronium ion (NO⁺) carrier, targets cysteine thiols (-SH) via electrophilic substitution, yielding S-nitrosothiol (SNO) and nitrite (NO₂⁻).
- Kinetic Features:
- Rapid kinetics (rate constant k ≈ 10⁷ M⁻¹s⁻¹).
- Favored in hydrophobic environments (e.g., lipid bilayers) or high-oxygen niches (e.g., lung epithelium).
Pathophysiological Contexts:
| Microenvironment | Target Protein/Residue | Functional Consequence |
|---|---|---|
| Inflammatory foci (iNOS↑) | p53 Cys124 | Apoptosis induction |
| Hyperoxic tissues | EGFR Cys residues | Pro-fibrotic signaling activation |
| Platelet membranes | CD36 | Thrombosis potentiation |
Small-Molecule Mediation via Glutathione (GSNO)
1. GSNO Biosynthesis and Equilibrium
- Formation: GSH + N2O3 -> GSNO + NO2- + H+
- Dynamic Equilibrium: GSNO + R-SH <=> R-SNO + GSH (K~eq~ ≈ 1-10)
The position of this equilibrium is influenced by the pKₐ of the acceptor thiol; thiols with lower pKₐ values exhibit a greater propensity for NO acceptance.
2. Enzymatic Control of S-Nitrosation
As the predominant intracellular nitric oxide (NO) carrier, S-nitrosoglutathione (GSNO) forms via electrophilic attack of NO on reduced glutathione (GSH + N2O3-> GSNO + NO2- + H+). Subsequent modification of target proteins occurs through thiol-S-nitrosothiol exchange (GSNO + R-SH <=> R-SNO + GSH). This reversible process is governed by the pKₐ of the receiving thiol (thiols with pKₐ ≤ 8 favor NO acceptance) and is enzymatically regulated:
- Glutathione transferase (GST): Catalyzes transnitrosation from GSNO to specific protein targets, exemplified by its activation of the H-Ras Cys118 residue, thereby initiating the MAPK signaling cascade.
- Thioredoxin (Trx): Reductively eliminates excessive SNO modifications, maintaining redox homeostasis (e.g., suppressing aberrant apoptosis signal-regulated kinase 1 (ASK1) activation).
Significantly, GSNO reductase (GSNOR) functions as a central negative regulator of systemic NO bioavailability by catabolizing GSNO (GSNO + NADH -> GSSG + NH3 + NAD+). GSNOR deficiency is implicated in pathologies, such as excessive airway NO accumulation in cystic fibrosis patients. Characterized by moderate kinetics (k ≈ 10 ms), this pathway underpins fundamental cytoplasmic and mitochondrial matrix NO signaling.
Services You May Be Interested In:
Protein-Protein Transnitrosation
This pathway critically enables intracellular communication, particularly governing cellular growth, differentiation, stress adaptation, and apoptotic processes. Its fundamental mechanism involves the targeted transfer of nitric oxide (NO) from donor proteins to acceptor proteins, establishing spatially defined signaling cascades.
1. Donor Protein S-Nitrosothiol Dissociation
Donor proteins carry reversible S-nitrosation modifications (SNO) at cysteine thiol groups (-SH). Transfer initiation requires cleavage of this SNO bond, achieved through:
- Oxidative Triggering: Cellular oxidants (e.g., reactive oxygen species) facilitate dissociation via redox reactions. Hydrogen peroxide (H₂O₂), for instance, enhances SNO lability, promoting NO release.
- Photolytic Cleavage: Light-sensitive proteins undergo SNO bond rupture upon specific wavelength exposure, liberating NO—a mechanism prevalent in photoregulated enzymes and signaling pathways.
These processes release NO in a transfer-competent state for subsequent signaling events.
2. Acceptor Protein Nitrosation Mechanisms
The nucleophilic thiolate (-S⁻) of acceptor proteins reacts with released NO⁺ equivalents via distinct routes:
- Direct Nucleophilic Attack: NO⁺ combines with the acceptor thiol (P₂-SH/P₂-S⁻), forming S-nitrosated protein (P₂-SNO). This direct reaction frequently modifies cytoplasmic and membrane-associated proteins (e.g., kinases, receptors) regulating signal transduction.
- Enzyme-Catalyzed Transfer: Protein disulfide isomerase (PDI) facilitates transnitrosation through a transient disulfide bond intermediate (P₁-S-S-P₂). This mechanism ensures transfer fidelity, prevents uncontrolled NO diffusion, and maintains spatiotemporal signaling precision.
3. Signal Cascade Network Integration
Beyond singular modifications, this pathway constructs complex signaling networks:
- Cellular Process Regulation: NO transfer directly modulates critical functions. Examples include: NO transfer to XIAP inhibiting apoptosis; S-nitrosation of neuronal ion channels or kinases altering excitability and synaptic plasticity.
- Homeostatic Maintenance: Reverse transnitrosation (e.g., via PDI) and denitrosylase activity prevent excessive S-nitrosation, preserving NO homeostasis. Dysregulation contributes to pathologies like neurodegeneration and cancer.
- Intercellular Communication: While NO itself is membrane-permeable, transnitrosation pathways can influence receptor modifications, facilitating coordinated multicellular responses. Secondary messengers often mediate these intercellular signaling events.
Metal/Heme-Mediated Nitrosation Pathways
Catalytic Mechanism via Metal Centers
Transition metals or heme groups facilitate nitrosation by forming nitrosyl complexes (M−NO), subsequently oxidizing cysteine thiolates. The process involves:
- Nitrosyl Complex Formation: Transition metals (Fe/Cu) bind NO through donation of d-orbital electrons into NO's π antibonding orbital.
- Metal-Ligand Configuration & Stability:
- Fe: Linear Fe−N−O geometry susceptible to O₂ oxidation → inactivation.
- Cu: Bent Cu−N−O configuration exhibits antioxidant properties → physiologically dominant.
- Thiol Oxidation and NO⁺ Transfer:
- [Mn+−NO]δ++R−S−−>R−SNO+M(n+1)+
- Essence: The metal center converts NO into an electrophilic NO⁺ equivalent, targeting deprotonated thiols (-S⁻).
Key Enzymatic Systems: Structure and Function
Cytochrome c (Cyt c) in Mitochondrial Regulation:
- Active Site: Heme iron coordinates with Cys17/Cys19.
- Nitrosation Cascade:
- NO binds heme iron → displaces heme ligand → liberates Cys17.
- [Fe−NO] oxidizes thiols of adjacent proteins (e.g., Cys139 of Rieske protein in Complex III).
- Physiological Significance:
- Low [NO•]: Reversible electron transport chain inhibition → ROS modulation.
- High [NO•]: Irreversible SNO modification → mitochondrial apoptosis induction.
Copper-Zinc Superoxide Dismutase (SOD1) Catalysis:
- Structural Basis: Copper active site forms coordination complex with Cys111.
- Reaction Pathway:
- SOD1−Cu++NO⋅−>[SOD1−Cu2+−NO]+
[SOD1−Cu2+−NO]++Prx−S−−>Prx−SNO+SOD1−Cu2+
- SOD1−Cu++NO⋅−>[SOD1−Cu2+−NO]+
- Function: Transfers NO signal to peroxiredoxin (Prx), regulating cellular H₂O₂ metabolism.
Heme-Assisted Nitrosation Mechanisms
1. Allosteric Regulation via Heme-NO Binding
Nitric oxide (NO) coordination to heme iron induces atomic displacement above the porphyrin plane, triggering protein conformational rearrangement. This structural shift exposes solvent-inaccessible cysteine residues, enabling subsequent nitrosation reactions.
2. Hemoglobin (Hb) Oxygen-Sensing Function
Heme-NO binding affinity and cysteine accessibility are conformationally gated:
- Deoxygenated (T) State: High-affinity NO binding facilitates transnitrosation to Cysβ93, forming S-nitrosohemoglobin (SNO-Hb).
- Oxygenated (R) State: Low-affinity NO binding permits direct oxidation to nitrate (NO₃⁻), preventing SNO formation.
3. SNO-Hb in Vascular Regulation
- Hypoxic Tissues: T-state Hb releases SNO, activating the soluble guanylate cyclase (sGC)-cGMP pathway to induce vasodilation.
- Allosteric Coupling Evidence: Oxygen binding to Hb α-subunits promotes SNO release from β-subunits (Cysβ93).
Alternative Nitrosation Mechanisms
Nitrate Reductase Pathway
Bacterial enzymes (e.g., oral microbiota) catalyze dietary nitrate (NO₃⁻) reduction to nitrite (NO₂⁻) and subsequently nitric oxide (NO•), enabling nitrosation. Mammalian systems exhibit analogous biochemistry:
- Oral Microbiome: Symbiotic flora reduce NO₃⁻ → NO₂⁻ → NO• via nitrate reductase.
- Mammalian Enzymes:
- Mitochondrial aldehyde oxidase (mAO): Reduces NO₂⁻ to NO• under hypoxic conditions.
- Xanthine oxidoreductase (XOR): Generates NO• during ischemic stress via substrate transformation.
Peroxynitrite (ONOO⁻) Pathway: Dual Functionality
The role of ONOO⁻ in nitrosation remains debated:
- Conventional Understanding: ONOO⁻ primarily induces tyrosine nitration (3-NT) or cysteine oxidation (-SOH/-SO₂H).
- Emerging Evidence: In glutathione (GSH)-rich environments, ONOO⁻ indirectly facilitates S-nitrosation.
References
- Fernando V, Zheng X, Walia Y, Sharma V, Letson J, Furuta S. "S-Nitrosylation: An Emerging Paradigm of Redox Signaling." Antioxidants (Basel). 2019 Sep 17;8(9):404. doi: 10.3390/antiox8090404
- Sharma V, Fernando V, Letson J, Walia Y, Zheng X, Fackelman D, Furuta S. "S-Nitrosylation in Tumor Microenvironment." Int J Mol Sci. 2021 Apr 27;22(9):4600. doi: 10.3390/ijms22094600
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP. "Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I." Nat Med. 2013 Jun;19(6):753-9. doi: 10.1038/nm.3212










