Protein phosphorylation entails the covalent attachment of a phosphate group (PO₄³⁻) to specific amino acid residues—primarily serine (Ser), threonine (Thr), and tyrosine (Tyr). This post-translational modification dynamically regulates protein conformation, stability, enzymatic activity, and intermolecular interactions. In this paper, the molecular mechanism of protein phosphorylation, the role of related enzymes, and its role in biological functions will be discussed in a panoramic way.
1. Chemical Principles and Core Mechanisms
As the central switch governing cellular signaling, phosphorylation represents a covalent modification process precisely controlled by kinase-phosphatase systems. This section integrates three fundamental dimensions: enzymatic mechanisms, structural dynamics, and regulatory networks.
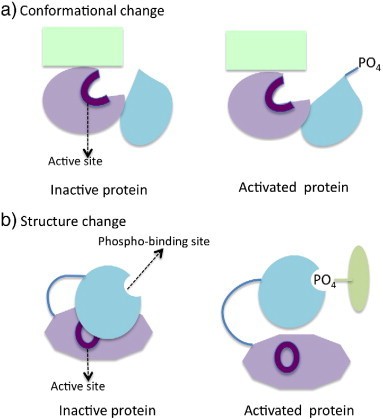 Mechanisms of modulating protein function by phosphorylation (Liu Y et al., 2014)
Mechanisms of modulating protein function by phosphorylation (Liu Y et al., 2014)
1.1 Core Enzymatic Machinery of Phosphorylation
| Enzyme Class | Catalytic Function | Recognition Motif | Biological Role |
|---|---|---|---|
| Ser/Thr Kinases (e.g., PKA) | γ-phosphate transfer to Ser/Thr | R-X-X-S/T | Metabolic/cell cycle regulation |
| Tyrosine Kinases (e.g., EGFR) | Tyr residue phosphorylation | SH2 domain binds pTyr | Growth factor signaling control |
| Dual-specificity (e.g., MEK1) | Concurrent Ser/Thr/Tyr modification | MAPK-docking site | MAPK cascade amplification |
| Phosphatases (PP1/PP2A/PTP) | Phosphate hydrolysis | Distinct Ser/Thr vs. Tyr specificity | Signal termination and homeostasis |
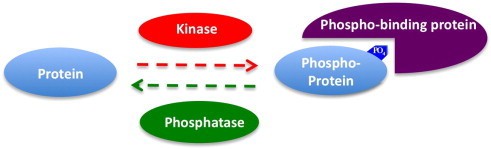 Relationships among kinase, phosphatase and phospho-binding protein (Liu Y et al., 2014)
Relationships among kinase, phosphatase and phospho-binding protein (Liu Y et al., 2014)
1.2 Structural Dynamics of Kinase Catalysis
Catalytic cycle exemplified by protein kinase A (PKA):
- Substrate Docking: Target peptide anchors to kinase's C-lobe via hydrophobic pockets.
- Nucleotide Binding: ATP adenine ring forms hydrogen bonds with conserved N-lobe lysine (Lys72).
- Phosphotransfer:
- Catalytic aspartate (Asp166) polarizes water molecules
- Nucleophilic attack on ATP γ-phosphate
- Phosphate transfer to substrate Ser/Thr hydroxyl
- Product Dissociation: ADP and phosphorylated peptide release from active site.
Allosteric Regulatory Hotspots:
- Activation Loop:
- Unphosphorylated state obstructs substrate access (e.g., Src kinase Tyr394)
- Phosphorylation induces conformational opening
- Phosphate-Binding Loop (P-loop): Gly-X-Gly-X-X-Gly motif coordinates ATP β/γ-phosphates
Select Service
Learn more
2.Phosphorylation Regulatory Networks
2.1 Signal Transduction Switching Mechanisms
Phosphorylation serves as a fundamental regulatory switch in cellular signaling, enabling precise responses to extracellular stimuli and intracellular demands through activation/inhibition cascades. Key pathways include:
MAPK Pathway:
Tertiary Kinase Phosphorylation Cascade: Extracellular stimuli initiate membrane receptor activation, triggering a sequential kinase cascade. MAP3Ks (e.g., Raf, TAK1), activated by upstream signals like Ras-GTP or TRAFs, phosphorylate and activate MAP2Ks (e.g., MEK1/2, MKK3/6). These bispecific MAP2Ks subsequently dually phosphorylate MAPKs (e.g., ERK, p38, JNK) at conserved threonine-tyrosine motifs. Activated MAPKs then phosphorylate downstream transcription factors (e.g., c-Myc, ATF2) and effector proteins (e.g., MNK1, MSK1).
Major MAPK Sub-pathways:
- ERK Pathway: Activated by growth factors (EGF, PDGF). Cascade: RAF → MEK1/2 → ERK1/2. Function: Cell proliferation/differentiation.
- p38 Pathway: Activated by stress (UV, inflammation, osmolarity). Cascade: TAK1 → MKK3/6 → p38. Function: Inflammation/apoptosis.
- JNK Pathway: Activated by stress (DNA damage, heat shock). Cascade: MEKK1 → MKK4/7 → JNK. Function: Apoptosis/metabolic regulation.
Core Regulatory Functions of Phosphorylation
- Signal Amplification & Digital Switching: MAPK activation requires dual phosphorylation within the T-X-Y motif (e.g., T-E-Y in ERK), creating a stringent gating mechanism. This enables a threshold response: low-intensity stimuli yield ineffective monophosphorylation, while high-intensity stimuli induce full activation via dual phosphorylation, generating a digital signal output.
- Spatiotemporal Precision: Scaffold proteins (e.g., KSR, JIP) localize kinases into microdomains, ensuring efficient cascade progression. Negative feedback is provided by phosphatases: MKPs dephosphorylate MAPK to terminate signaling, while PP2A acts on MAP3Ks/MAP2Ks to prevent excessive activation.
- Cross-pathway Integration: Phosphorylation enables crosstalk:
- AKT phosphorylates Raf(S259), inhibiting the ERK pathway (contributing to tumor drug resistance).
- PKC phosphorylates Raf(S338), promoting ERK activation (facilitating GPCR signal integration).
PI3K/AKT Pathway:
- Core Signaling Components & Phosphorylation Cascade: Growth factors initiate signaling by activating receptor tyrosine kinases (RTKs). This triggers PI3K, which phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP₂) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP₃). PIP₃ recruits AKT to the plasma membrane via its PH domain, enabling PDK1 to phosphorylate AKT at Thr308 (T308). Subsequent phosphorylation at Ser473 (S473) by mTORC2 fully activates AKT. The fully activated kinase then translocates to subcellular compartments to phosphorylate numerous downstream effector proteins.
- PI3K (Phosphatidylinositol 3-Kinase): Activated by RTKs (e.g., IGF-1R) or GPCRs; catalyzes PIP₂ to PIP₃ conversion.
- AKT (Protein Kinase B): Requires dual phosphorylation for full activation: PDK1 targets T308 (activation loop), conferring partial activity; mTORC2 phosphorylates S473 (hydrophobic motif), enabling maximal catalytic function. Activated AKT phosphorylates over 100 substrates.
- Principal Downstream Effector Pathways & Biological Outcomes:
AKT-mediated phosphorylation of key substrates drives critical cellular functions:- Metabolic Regulation: Phosphorylation inhibits GSK3β and TSC2, promoting glycogen synthesis and suppressing autophagy.
- Anti-Apoptosis: Phosphorylation inactivates pro-apoptotic proteins BAD and FOXO (via inactivation/nuclear export), enhancing cell survival.
- Proliferation/Growth: Phosphorylation activates mTORC1 and MDM2, stimulating protein synthesis, inhibiting p53, and accelerating cell cycle progression.
Essential Regulatory Functions of Phosphorylation
- Signal Initiation Precision & Amplification:
- Membrane Recruitment Switch: PIP₃ binding to AKT's PH domain localizes AKT to the membrane, exposing T308 for PDK1 phosphorylation.
- Dual Phosphorylation Synergy: T308-P increases substrate affinity; S473-P enhances catalytic efficiency. Combined phosphorylation boosts AKT activity ~100-fold.
- Negative Feedback Control:
- PTEN (Phosphatase): Antagonizes PI3K by dephosphorylating PIP₃ to PIP₂, terminating the signal (PTEN is a key tumor suppressor, mutated in ~40% of endometrial cancers).
- mTORC1/S6K Pathway Feedback: S6K phosphorylates IRS1, impairing its ability to link PI3K to activated RTKs, contributing to insulin resistance.
JAK-STAT Pathway:
Core Pathway Components & Activation Mechanism
- Signal Transduction Cascade: Cytokine binding induces receptor dimerization, triggering mutual JAK transphosphorylation and activation. Activated JAKs phosphorylate receptor tyrosine residues, enabling STAT recruitment and phosphorylation. Phosphorylated STATs dimerize and translocate to the nucleus to regulate target gene transcription.
- JAK (Janus Kinase): Includes isoforms JAK1, JAK2, JAK3, TYK2 (non-receptor tyrosine kinases). Constitutively bound to receptor intracellular domains. Receptor dimerization prompts JAK autophosphorylation at conserved tyrosine sites (e.g., JAK2 Tyr1007), achieving full activation.
- STAT (Signal Transducer and Activator of Transcription): Possess SH2 domains that bind receptor phosphotyrosines (e.g., IFNγ receptor pTyr440). JAK-mediated phosphorylation of STAT C-terminal tyrosine residues (e.g., STAT1 Tyr701) induces conformational changes. This drives phosphotyrosine-SH2-dependent dimer formation and nuclear translocation.
- STAT Dimer Functions:
Specific STAT dimers mediate distinct cytokine responses:- STAT1: Activated by IFNγ/IFNα/β. Target genes: IRF1, MHC-II. Functions: Antiviral defense, immune activation.
- STAT3: Activated by IL-6/IL-10. Target genes: Bcl-2, VEGF. Functions: Anti-apoptosis, tumor promotion.
- STAT5: Activated by erythropoietin. Target genes: Cyclin D1, BIRC5. Functions: Hematopoietic regulation, proliferation.
Essential Regulatory Roles of Phosphorylation
- Precision Switching Mechanisms:
- JAK Autophosphorylation: Receptor dimerization relieves autoinhibition, enabling JAK transphosphorylation (e.g., JAK2 Tyr1007). This exposes the kinase active site.
- STAT Phosphorylation: Phosphorylation (e.g., STAT1 Tyr701) enables intramolecular SH2-pTyr binding, releasing autoinhibitory constraints and exposing the DNA-binding domain. This facilitates STAT dimerization via reciprocal SH2-pTyr interactions.
- Negative Feedback Regulation:
- SOCS Proteins (Suppressors of Cytokine Signaling): STAT-induced expression. Inhibit signaling by: 1) Competing with receptor/JAK binding sites, blocking phosphorylation; 2) Recruiting E3 ubiquitin ligases to target JAKs for degradation (e.g., SOCS1-mediated JAK2 ubiquitination).
- SHP Phosphatases: Terminate signals by dephosphorylating activated receptors or JAK kinases.
2.2 Cell Cycle Engine Control
Precise cell division timing is orchestrated by Cyclin-CDK complexes through stage-specific phosphorylation:
- G1/S Transition: Cyclin D-CDK4/6 phosphorylates retinoblastoma protein (Rb) at Ser780/Ser795, liberating E2F transcription factors to activate DNA replication genes—essential for S-phase commitment.
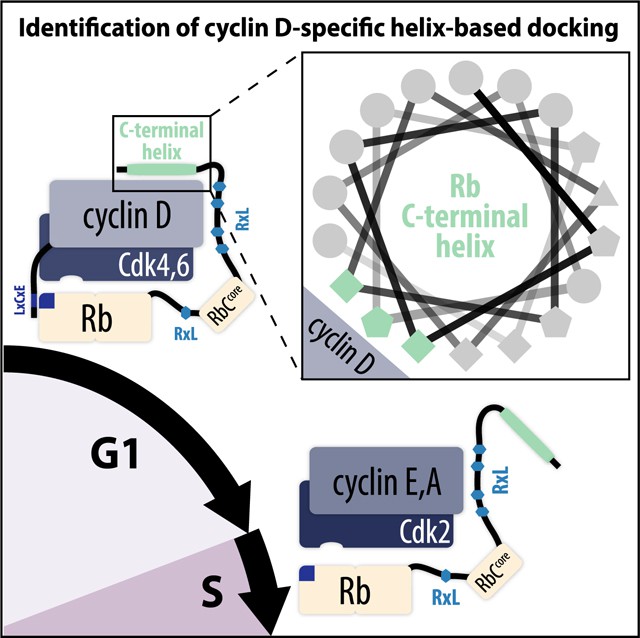 Cyclin D binds to CDK4/6 and promotes Rb phosphorylation as cells move from G1 to S phase (Topacio BR et al., 2019)
Cyclin D binds to CDK4/6 and promotes Rb phosphorylation as cells move from G1 to S phase (Topacio BR et al., 2019)
- M-Phase Initiation: Cyclin B-CDK1 phosphorylates nuclear lamins (e.g., Ser22), triggering nuclear envelope breakdown and enabling chromosomal segregation during mitosis.
2.3 Metabolic Pathway Dynamics
Cellular energy homeostasis and nutrient responsiveness are phosphorylation-regulated:
- Insulin Signaling: Insulin receptor autophosphorylation → IRS1 (Tyr612) phosphorylation → PI3K/AKT activation → AS160 (Thr642) phosphorylation → GLUT4 vesicle membrane fusion → Enhanced glucose uptake. Dysregulation contributes to diabetic pathogenesis.
- AMPK Energy Sensing: Low energy (AMP/ADP) → LKB1-mediated AMPK activation → ACC (Ser79) phosphorylation → Malonyl-CoA reduction → Enhanced fatty acid oxidation. This pathway restores energy balance during nutrient deprivation or exercise.
3.Advanced Phosphorylation Regulatory Paradigms
3.1 Signal Amplification via Phosphorylation Cascades: cGAS-STING Spatiotemporal Control
Molecular Mechanism Refinement:
- TBK1 Hierarchical Activation: STING recruits TBK1 → TBK1 autophosphorylation at Tyr172 enables full activation → Activated TBK1 phosphorylates STING Ser366 → Positive feedback loop established
- IRF3 Synergistic Modification: CK1ε phosphorylates IRF3 C-terminal residues (Ser396/398/402/404) → Enhances dimer stability beyond Ser386 phosphorylation
- Negative Regulation:
- USP20 deubiquitinates STING → Sustains signaling duration
- SHP2 dephosphorylates TBK1 Tyr239 → Signal attenuation
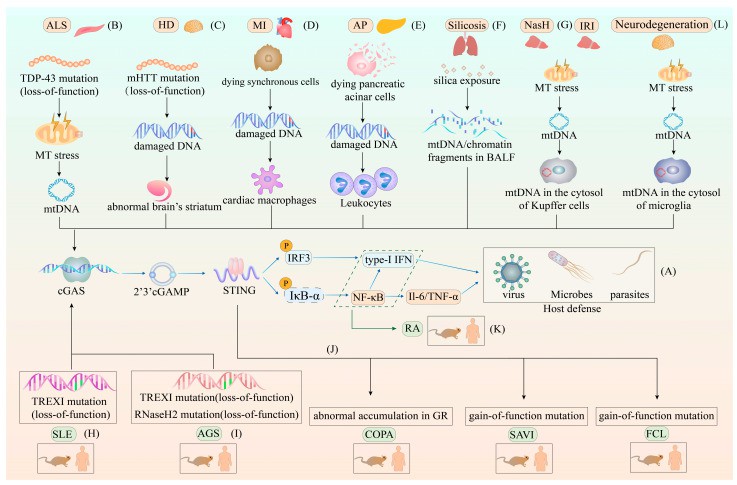 The different roles of the cGAS–STING pathway in host defense and various diseases(Zhou J et al., 2023)
The different roles of the cGAS–STING pathway in host defense and various diseases(Zhou J et al., 2023)
Physiopathological Significance:
- Antiviral immunity: >1000-fold signal amplification within 5 minutes
- Autoimmunity: STING S366A mutation causes lupus-like pathology (murine models)
3.2 Phosphocode Complexity Expansion
(1) Tau Protein Pathological Network
Disease-Specific Phosphosignatures:
- Alzheimer's: pSer202/pThr205/pSer404 (AT8 epitope) → Microtubule disassembly → Neurofibrillary tangles
- Progressive Supranuclear Palsy: pSer409/pSer422 (PG5/AP422) → Spherical inclusion formation
- Corticobasal Degeneration: pThr231/pSer235 (AT180) → Neuronal vacuolization
Regulatory Crosstalk:
- Kinase Synergy: GSK3β pSer202 → CDK5 pThr205 → MARK pSer262
- Phosphosite Antagonism: PKA-mediated pSer214 inhibits CDK5-driven pSer422 pathology
(2) 14-3-3 Phosphodependent Recognition
Structural Basis:
Amphipathic groove binds pSer/pThr-containing motifs:
- Mode I: Arg-Ser-X-pSer-X-Pro (e.g., Bad pSer112)
- Mode II: Arg-X-X-pSer-X-Pro (e.g., Cdc25 pSer216)
Functional Diversity:
- Bad pSer112: Masks BH3 domain → Anti-apoptotic
- CFTR pSer768: Enhances membrane localization
- KSR1 pSer392: Scaffolds MAPK signaling
3.3 Subcellular Localization Machinery
(1) Nucleocytoplasmic Shuttling: NF-κB Allostery
Phosphorylation-Driven Conformational Switching:
- Basal state: IκBα masks NLS, exposes NES → Cytoplasmic retention
- Activation:
- IKKβ pSer536 → IκBα dissociation/degradation
- CK2 pSer529 → NLS exposure
- Nuclear Retention: pSer536 disrupts NES conformation → Impedes CRM1 binding (Leu-X-X-Leu-Leu motif)
(2) Spatial Signal Organization: EGFR Paradigm
- Phosphodependent Adaptor Recruitment:
- pTyr1068: GRB2-SH2 → RAS-MAPK activation
- pTyr1086: SHC-PTB → PI3K-Akt initiation
- pTyr1045: Cbl-E3 ubiquitin ligase → Receptor degradation
- Signal Polarization: Frontal EGFR phosphorylation → Localized PI3K recruitment → PIP₃ gradient formation → Directed cell migration
4. Advanced Detection Technologies and Analytical Innovations
4.1 Phosphoproteomic Standardization: Optimization and Challenges
Sample Preparation Critical Factors:
- Phosphatase Inhibition Cocktail:
- Broad-spectrum: NaF (10 mM) + β-glycerophosphate (20 mM)
- Tyrosine-specific: Sodium vanadate (1 mM)
- Denaturation Efficiency: 8M urea lysis outperforms SDS (preserves enzymatic digestibility)
Peptide Enrichment Method Comparison:
| Technique | Principle | Advantages | Limitations |
|---|---|---|---|
| TiO₂ | Acidic pH (2.5) Ti⁴⁺-phosphate chelation | High specificity (>90%) | Acidic peptide loss |
| IMAC (Fe³⁺/Ga³⁺) | Metal ion-phosphate coordination | Effective pTyr enrichment | EDTA interference |
| Metal Oxides (MoO₃/MgO) | Enhanced phosphate affinity | Salt-tolerant | Batch variability |
Mass Spectrometry Advancements:
- Fragmentation Selection:
- HCD: Generates complete b/y ions → Optimal for quantification
- ETD: Preserves labile modifications → Superior for multisite analysis
- Quantification Strategies:
- TMT: 11-plex capability → Requires reporter ion compression correction
- Label-free (LFQ): MaxLFQ algorithm → Normalizes cross-sample variation
Data Analysis Breakthroughs:
- Site Localization:
- Ascore: Fragment ion probability scoring (δ-score >19; p<0.01)
- PhosphoRS: Isotopic pattern verification → Reduces false positives
- Pathway Integration:
- PhosR: Kinase-substrate network reconstruction
- KSEA: Upstream kinase activity inference from phosphosite dynamics
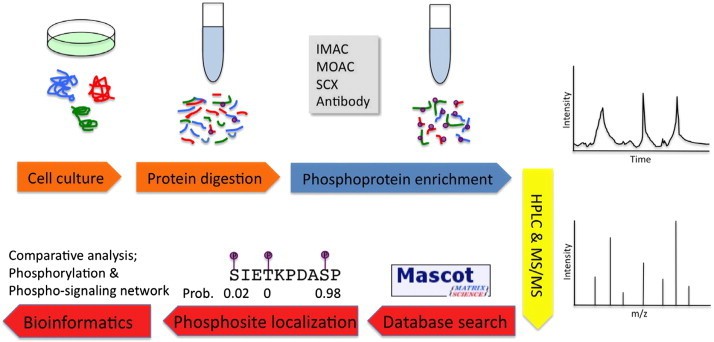 Summary of MS-based phosphoproteomics experiments (Liu Y et al., 2014)
Summary of MS-based phosphoproteomics experiments (Liu Y et al., 2014)
4.2 Single-Cell Resolution: Multidimensional Advances
Mass Cytometry (CyTOF) Upgrades:
- Detection System:
- Lanthanide-tagged antibodies (Eu-Lu) → Zero spectral overlap
- Throughput: >40 phosphosignaling proteins/cell
- Computational Analysis:
- SPADE: Cluster-based visualization (e.g., PD-1 pTyr248 gradients in T-cell subsets)
- Diffusion Map: Pseudotemporal trajectory modeling (e.g., pSyk dynamics during B-cell differentiation)
Satial Phosphoproteomics:
- CODEX: Antibody-DNA conjugates → Cyclic imaging → Tissue microniche phosphosignaling mapping
- MIBI-TOF: Ion beam mass spectrometry → Spatial heterogeneity mapping (e.g., pERK in breast cancer niches)
To learn more about phosphoproteomics, please refer to "Phosphoproteomics in Cancer Research: A Data-Driven Approach".
Functional Diversity of Protein Phosphorylation
1. Signal Transduction: Molecular Switch Regulation
- Cascade Amplification: This study identifies the EGFR-MAPK phosphorylation cascade as a pivotal regulatory switch in SARS-CoV-2 pathogenesis. Viral spike protein binding activates EGFR, inducing phosphorylation at critical intracellular tyrosine residues (Y1068/Y1173). This modification promotes EGFR-ACE2 complex formation with bidirectional consequences: ACE2 membrane enrichment enhances viral attachment, while upregulated EGFR endocytic activation facilitates viral entry. Subsequently, EGFR phosphorylation initiates downstream MAPK signaling, driving sequential RAS→RAF→MEK→ERK phosphorylation that remodels the cytoskeleton to enable viral endocytosis and intracellular trafficking. Crucially, pharmacological inhibition targeting EGFR phosphorylation (e.g., osimertinib) or MEK phosphorylation (e.g., trametinib) significantly reduces both pseudovirus and live-virus infection rates, validating this phosphorylation network as a promising antiviral therapeutic target (Engler M et al., 2023).
- Rapid Signal Termination: The DNA damage response (DDR) employs a specific mechanism to inhibit viral replication: phosphorylation of the evolutionarily conserved T518 residue within the Simian Virus 40 large T antigen (LT). This molecular switch, functionally validated using the phosphomimetic T518D mutation, directly impedes LT helicase activity on extended DNA templates (while minimally affecting short templates) and halts replication fork progression. Crucially, this regulatory event precisely targets LT's replicative function without compromising its transcriptional activation capabilities or protein interaction networks. Through this phosphorylation-dependent molecular brake, DDR kinases selectively terminate viral genome duplication, constituting a crucial host defense strategy against viral infection (Homiski C et al., 2024).
2. Cell Cycle: Temporal Control System
- Phase Transition Mechanisms: Rb phosphorylation mediated by CDK4/6 starts the cell cycle process, and the positive feedback loop of CDK2-Rb-E2F forms a bistable switch through phosphorylation cascade, which triggers irreversible G1/S conversion when the activity of CDK2 exceeds the threshold, ensuring the key decision point of cell cycle setting before DNA replication (Kim S et al., 2022).
- Checkpoint Enforcement: Following DNA damage, ATM kinase targets E6AP at Ser218, alleviating its ubiquitin-mediated degradation of MASTL. Stabilized MASTL subsequently inhibits the PP2A/B55 phosphatase complex. This inhibition sustains phosphorylation of CDK substrates, thereby facilitating cell cycle re-entry. Collectively, this ATM-E6AP-MASTL-PP2A/B55 signaling axis constitutes a molecular "damage timer," establishing the transient nature of checkpoint arrest (Li Y et al., 2023).
3. Transcriptional Regulation
- p53: ZNF498 drives hepatocellular carcinoma (HCC) progression by suppressing p53-mediated cell death. It competes with the PKCδ/p53INP1 complex for p53 binding, thereby preventing Ser46 phosphorylation. This blockade inactivates p53's transcriptional function, inhibiting both apoptosis and ferroptosis. Critically, phosphorylation at p53 Ser46 serves as a molecular switch governing cell death fate; its disruption by ZNF498 represents a core mechanism for inactivating non-mutant p53 in HCC (Zhang X et al., 2022).
4. Protein Interactions: Conformational Switching
- Scaffold-Mediated Recognition: β-arrestin achieves non-canonical activation by binding instantaneously to core domains of the GPCR (rather than C-terminal tails), disrupting critical inter-domain electrostatic networks (e.g., R169-E191/K295). This interaction induces conformational changes that trap β-arrestin at the plasma membrane. Subsequent phosphorylation-mediated localization occurs via two distinct pathways: classical phosphorylation of the GPCR tail stabilizes GPCR/β-arrestin scaffold formation, while lipid-directed recruitment involves β-arrestin binding phosphorylated inositol phospholipids (PIP₂), enriching it at clathrin-coated structures (CCS). Localization to CCS involves direct interactions with lattice components like clathrin and AP2, driving molecular aggregation. This aggregation yields divergent functional outcomes: CCS-aggregated β-arrestin independently activates ERK signaling (GPCR-independent), while stable scaffold complexes facilitate classical downstream GPCR signaling (Eichel K et al., 2018).
- Phase Separation Control: CK1δ/ε-mediated multisite phosphorylation of FUS acts as a "conformational stability switch," preventing its pathological transition from a functional RNA-binding protein to a neurotoxic entity. This post-translational modification enhances FUS solubility, suppresses aberrant aggregation, and inhibits stress granule assembly. Consequently, FUS-mediated neurotoxicity in neurodegenerative diseases is alleviated (Kishino Y et al., 2022).
References
- Engler M, Albers D, Von Maltitz P, Groß R, Münch J, Cirstea IC. "ACE2-EGFR-MAPK signaling contributes to SARS-CoV-2 infection." Life Sci Alliance. 2023 Jul 4;6(9):e202201880. doi: 10.26508/lsa.202201880
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, von Zastrow M. "Catalytic activation of β-arrestin by GPCRs." Nature. 2018 May;557(7705):381-386. doi: 10.1038/s41586-018-0079-1
- Homiski C, Dey-Rao R, Shen S, Qu J, Melendy T. "DNA damage-induced phosphorylation of a replicative DNA helicase results in inhibition of DNA replication through attenuation of helicase function." Nucleic Acids Res. 2024 Sep 23;52(17):10311-10328. doi: 10.1093/nar/gkae663
- Kim S, Leong A, Kim M, Yang HW. "CDK4/6 initiates Rb inactivation and CDK2 activity coordinates cell-cycle commitment and G1/S transition." Sci Rep. 2022 Oct 7;12(1):16810. doi: 10.1038/s41598-022-20769-5
- Li Y, Wang F, Li X, Wang L, Yang Z, You Z, Peng A. "The ATM-E6AP-MASTL axis mediates DNA damage checkpoint recovery." Elife. 2023 Sep 6;12:RP86976. doi: 10.7554/eLife.86976
- Zhang X, Zheng Q, Yue X, Yuan Z, Ling J, Yuan Y, Liang Y, Sun A, Liu Y, Li H, Xu K, He F, Wang J, Wu J, Zhao C, Tian C. "ZNF498 promotes hepatocellular carcinogenesis by suppressing p53-mediated apoptosis and ferroptosis via the attenuation of p53 Ser46 phosphorylation." J Exp Clin Cancer Res. 2022 Feb 28;41(1):79. doi: 10.1186/s13046-022-02288-3
- Liu Y, Chance MR. "Integrating phosphoproteomics in systems biology." Comput Struct Biotechnol J. 2014 Aug 1;10(17):90-7. doi: 10.1016/j.csbj.2014.07.003
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, Sage J, Kõivomägi M, Skotheim JM. "Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein's C-Terminal Helix." Mol Cell. 2019 May 16;74(4):758-770.e4. doi: 10.1016/j.molcel.2019.03.020
- Zhou J, Zhuang Z, Li J, Feng Z. "Significance of the cGAS-STING Pathway in Health and Disease." Int J Mol Sci. 2023 Aug 28;24(17):13316. doi: 10.3390/ijms241713316
- Kishino Y, Matsukawa K, Matsumoto T, Miyazaki R, Wakabayashi T, Nonaka T, Kametani F, Hasegawa M, Hashimoto T, Iwatsubo T. "Casein kinase 1δ/ε phosphorylates fused in sarcoma (FUS) and ameliorates FUS-mediated neurodegeneration." J Biol Chem. 2022 Aug;298(8):102191. doi: 10.1016/j.jbc.2022.102191











