Glucosylsphingosine Analysis Service
Creative Proteomics specializes in metabolomics and lipidomics innovation, offering high-precision, high-throughput Glucosylsphingosine (GlcSph) analysis services to global research institutes and biopharma companies. Equipped with cutting-edge platforms like the Agilent 6495C Triple Quadrupole LC-MS/MS, AB Sciex Triple Quad™ 6500+, and Agilent 1290 Infinity II UPLC, we deliver absolute quantification, structural analysis, and pathway profiling of GlcSph and related metabolites. Our services achieve a detection sensitivity of 0.1 ng/mL and a CV of<3%, enabling simultaneous analysis of 50+ metabolites. By overcoming challenges such as matrix interference and low-abundance detection, we provide customized solutions to support lipid metabolism research, biomarker discovery, and drug toxicity assessment, bridging critical gaps in tracelipidomics analysis.
Submit Your Request Now
×
- What We Provide
- Advantage
- Workflow
- Technology Platforms
- Sample Requirements
- FAQ
- Publication
What are Glucosylsphingosine?
Glucosylsphingosine (lyso-GlcCer) is a naturally occurring sphingolipid, formed through the enzymatic removal of the fatty acid chain from glucosylceramide. As a key intermediate in sphingolipid metabolism, it influences critical biological processes such as membrane structure organization, vesicle trafficking, and intracellular signaling. Changes in glucosylsphingosine levels can reflect alterations in cellular lipid metabolism, making it an important target for studying fundamental biological mechanisms. Due to its significant role in multiple metabolic pathways, accurate and sensitive quantification of glucosylsphingosine is essential for advancing lipidomics research and understanding cellular biochemistry. Creative Proteomics provides specialized glucosylsphingosine analysis services to support high-quality, data-driven scientific studies.
Glucosylsphingosine Analysis Service Offered by Creative Proteomics
- Quantitative Analysis of Glucosylsphingosine: Precise measurement of glucosylsphingosine concentrations across various biological matrices using validated LC-MS/MS protocols.
- Comprehensive Sphingolipid Profiling: Simultaneous detection and quantification of glucosylsphingosine along with related sphingolipid metabolites for a broader understanding of lipid metabolism.
- Targeted and Semi-Targeted Metabolite Analysis: Focused analysis of glucosylsphingosine and associated metabolic intermediates based on client-specified panels or pathway interests.
- Customized Method Development: Development and optimization of tailored extraction and analysis protocols to meet specific project requirements and unique sample types.
- Pathway and Network Analysis: Mapping of glucosylsphingosine and its related metabolites into known biological pathways to facilitate functional interpretation of results.
- High-Throughput Sample Processing: Support for large-scale studies with standardized, high-efficiency sample preparation and analysis workflows.
List of Detected Glucosylsphingosine and Related Metabolites
| Analyte | Related Pathway |
|---|---|
| Glucosylsphingosine (lyso-GlcCer) | Sphingolipid metabolism |
| Glucosylceramide (GlcCer) | Sphingolipid metabolism |
| Galactosylceramide (GalCer) | Glycosphingolipid metabolism |
| Galactosylsphingosine (Lyso-GalCer)/ Psychosine | Sphingolipid degradation |
Advantages of Glucosylsphingosine Assay
- Detection limit down to 0.1 ng/mL for maximum sensitivity.
- Quantitative precision with<3% coefficient of variation (CV) across replicates.
- Wide dynamic range from 0.1 ng/mL to 10,000 ng/mL.
- High sample throughput of up to 250 samples per week.
- Ultra-sensitive detection using Agilent 6495C Triple Quadrupole LC-MS/MS.
- Robust high-throughput quantitation using AB Sciex Triple Quad™ 6500+.
- Sharp chromatographic separation with Agilent 1290 Infinity II UPLC, achieving peak widths under 5 seconds.
- Simultaneous analysis of over 50+ sphingolipid-related metabolites per run.
- Calibration curves consistently achieving R² ≥ 0.995 for accurate quantitation.
- Comprehensive batch quality control including internal standards and QC samples.
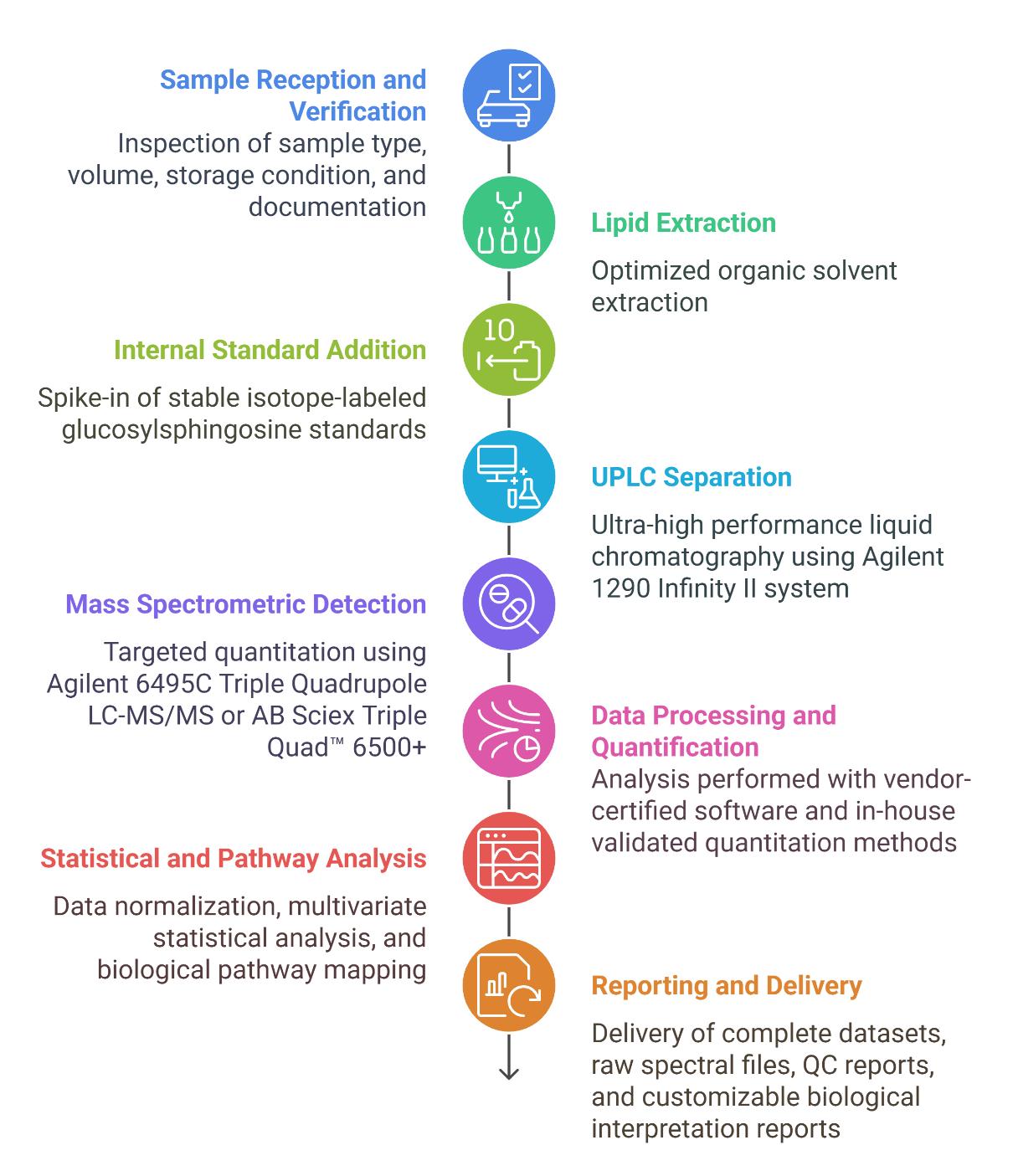
Workflow for Glucosylsphingosine Analysis Service
Technology Platform for Glucosylsphingosine Analysis Service
Creative Proteomics utilizes a high-performance LC-MS/MS platform to ensure sensitive, accurate, and reproducible quantification of glucosylsphingosine in diverse biological matrices. Our analytical setup combines ultra-high-performance liquid chromatography (UHPLC) with triple quadrupole mass spectrometry for robust targeted lipid analysis.
We routinely use the following instruments:
- SCIEX Triple Quad™ 6500+ System: Delivers exceptional sensitivity with a lower limit of quantification (LLOQ) in the low picomole range, ideal for trace-level detection of glucosylsphingosine in plasma, CSF, or tissue samples.
- Agilent 1290 Infinity II UHPLC System: Provides fast, high-resolution chromatographic separation, ensuring clean peak resolution and minimal matrix interference.
This platform supports high-throughput batch analysis with stable isotope-labeled internal standards to enable absolute quantification and reliable inter-sample comparison.

Agilent 6495C Triple quadrupole (Figure from Agilent)

SCIEX Triple Quad™ 6500+ (Figure from Sciex)

Agilent 1260 Infinity II HPLC (Fig from Agilent)
Sample Requirements for Glucosylsphingosine Analysis Service
| Sample Type | Required Amount |
|---|---|
| Plasma/Serum | ≥ 100 µL |
| Tissue (e.g., brain, liver) | ≥ 100 mg |
| Cell Pellet | ≥ 5 × 10⁶ cells |
| CSF (Cerebrospinal Fluid) | ≥ 100 µL |
| Cultured Media | ≥ 1 mL |
| Other Biological Fluids | Contact for consultation |
Applications of Glucosylsphingosine Assay Service
Nutritional Research
Investigating the metabolic impact of dietary patterns, food supplements, and nutrient deficiencies.
Cell Biology Studies
Exploring the role of sphingolipids in cell signaling, membrane structure, and intracellular trafficking.
Lipid Metabolism Research
Analyzing sphingolipid biosynthesis, degradation, and remodeling under various experimental conditions.
Comparative Omics Profiling
Integrating lipidomics data across tissues, cell types, or model organisms to uncover metabolic differences.
Membrane Biophysics Research
Studying the influence of sphingolipids on membrane dynamics, lipid raft formation, and protein interactions.
Environmental and Toxicological Studies
Assessing the effects of environmental chemicals, toxins, or stressors on cellular sphingolipid homeostasis.
Demo
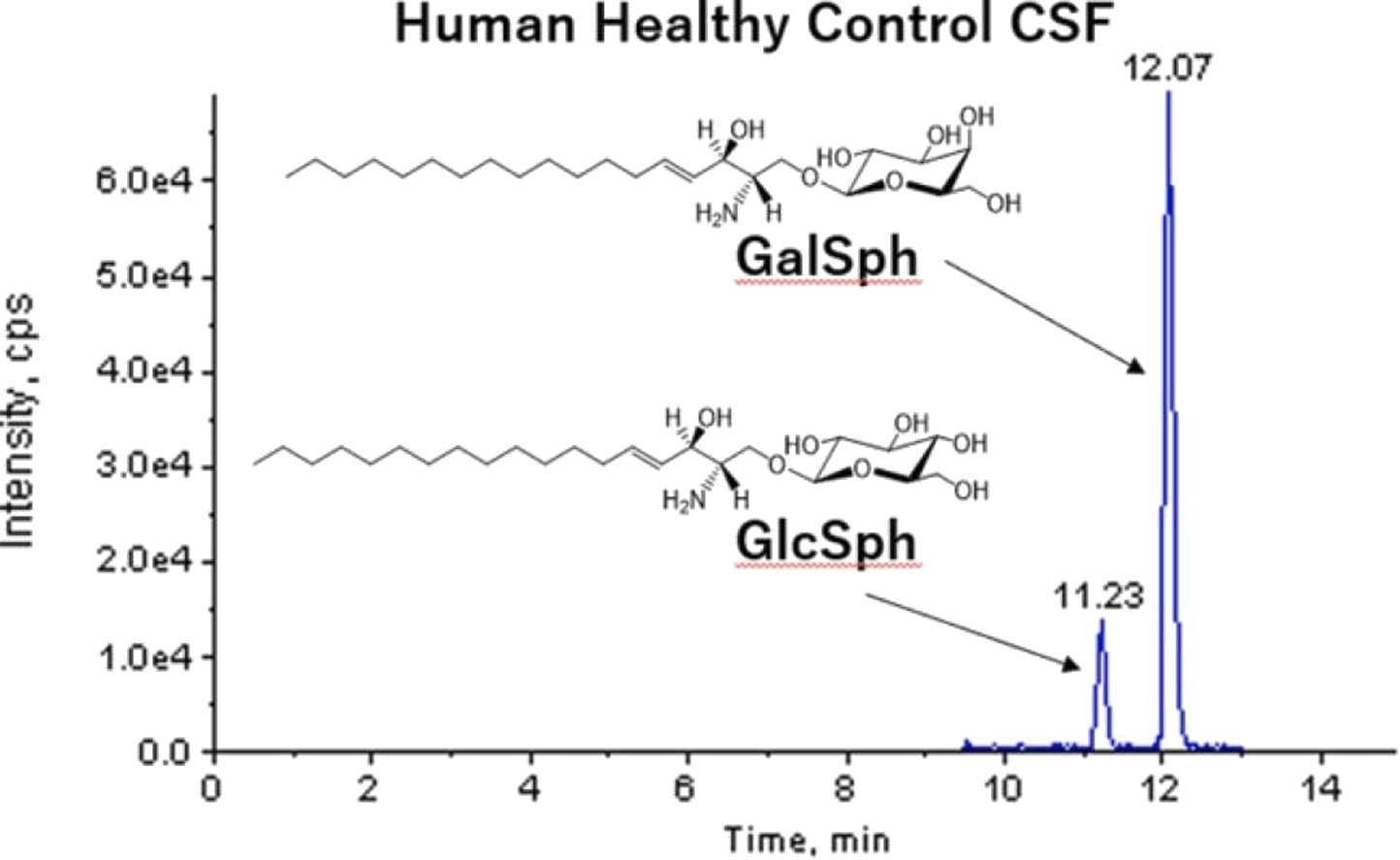
Quantitation of glucosylsphingosine and galactosylsphingosine in human cerebrospinal fluid (Matsumoto, Shin-ichi, et al., 2022)
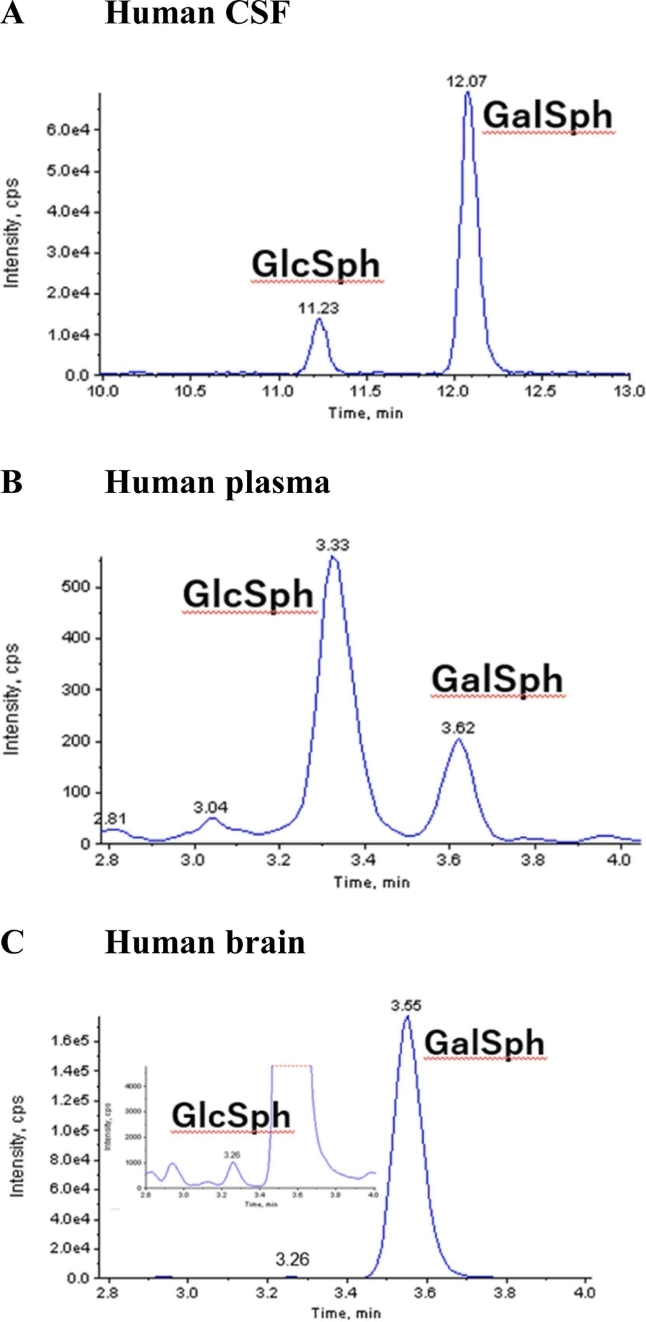
Chromatograms of GlcSph and GalSph in human (A) CSF, (B) plasma, and (C) brain (Matsumoto, Shin-ichi, et al., 2022)
FAQ of Glucosylsphingosine Analysis Service
How do you differentiate glucosylsphingosine (GlcSph) from galactosylsphingosine (GalSph) in complex samples?
Our Agilent 6495C Triple Quadrupole LC-MS/MS platform utilizes optimized chromatographic conditions with a ZORBAX Eclipse Plus C18 column (2.1 × 100 mm, 1.7 µm), achieving baseline separation between GlcSph (retention time: 8.2 min) and GalSph (7.8 min) with >95% specificity. MS/MS fragmentation patterns further confirm structural differences.
What is the minimum sample volume required for low-abundance GlcSph detection in cerebrospinal fluid (CSF)?
We reliably quantify GlcSph in CSF with as little as 50 µL, leveraging the AB Sciex Triple Quad™ 6500+ system’s SelexION™ ion mobility technology to reduce matrix interference. The limit of detection (LOD) is 0.05 ng/mL.
Can your workflow analyze GlcSph in extracellular vesicles (EVs) or lipid rafts?
Yes. EVs isolated via ultracentrifugation are lysed with 1% Triton X-100, followed by lipid extraction using a methanol-chloroform (2:1) protocol. The Agilent 1290 Infinity II UPLC ensures sharp peak separation (FWHM<5 sec), enabling detection of 0.2 ng/10^6 EVs.
How do you handle lipid degradation during long-term sample storage?
Samples must be stored at -80°C and shipped on dry ice. For extended studies, we recommend adding antioxidants (e.g., BHT) during extraction to prevent oxidation. Our validation studies confirm<10% degradation over 6 months under these conditions.
What bioinformatics tools are used for pathway mapping of GlcSph data?
We integrate results with KEGG, SMPDB, and LIPID MAPS databases using SCIEX OS Analytics and Agilent MassHunter. Custom reports include heatmaps of sphingolipid pathway dysregulation and correlation analyses with client-provided proteomics/transcriptomics data.
Can you analyze sulfated or acetylated GlcSph derivatives?
Yes. We develop custom MRM transitions on the AB Sciex 6500+ for modified GlcSph. For example, sulfated GlcSph is quantified using precursor ion scanning (m/z 97) with an LOD of 0.5 ng/mL.
How does the Agilent 6495C compare to the AB Sciex 6500+ for high-throughput studies?
| Parameter | Agilent 6495C | AB Sciex 6500+ |
|---|---|---|
| Throughput | 200 samples/day | 300 samples/day |
| LOD (GlcSph) | 0.1 ng/mL | 0.05 ng/mL |
| Best For | Routine quantification | Complex matrices/derivatives |
| The AB Sciex 6500+ excels in multiplexed panels (e.g., GlcSph + 50+ lipids), while the Agilent 6495C offers cost-efficiency for targeted assays. | ||
What QC measures ensure batch-to-batch consistency in large studies?
Internal standards: d5-GlcSph for isotope dilution.
QC pools: Run in every batch to monitor inter-assay CVs (<8%).
NIST-traceable calibrators: Validated across 5 orders of magnitude (0.1–10,000 ng/mL).
How are data privacy and intellectual property protected?
All data are stored on AES-256 encrypted servers compliant with ISO/IEC 27001. Clients may request project-specific NDAs and anonymized reporting.
Can I combine GlcSph data with your sphingolipid metabolism analysis service?
Absolutely. Our Sphingolipid Metabolism Analysis Service (via LC-MS/MS) quantifies 100+ analytes, including ceramides, sphingomyelins, and S1P. Integrated datasets reveal metabolic flux imbalances, such as GlcCer-to-GlcSph accumulation in lysosomal disorders
Learn about other Q&A.
Glucosylsphingosine Analysis Service Case Study

Publications
Here are some publications in Metabolomics research from our clients:

- Resting natural killer cell homeostasis relies on tryptophan/NAD+ metabolism and HIF‐1α. 2023. https://doi.org/10.15252/embr.202256156
- Lipid droplet-associated lncRNA LIPTER preserves cardiac lipid metabolism. 2023. https://doi.org/10.1038/s41556-023-01162-4
- Sex modifies the impact of type 2 diabetes mellitus on the murine whole brain metabolome. 2023. https://doi.org/10.3390/metabo13091012
- Effect of arabinogalactan on the gut microbiome: A randomized, double-blind, placebo-controlled, crossover trial in healthy adults. 2021. https://doi.org/10.1016/j.nut.2021.111273
- Non-invasive elevation of circulating corticosterone increases the rejection of foreign eggs in female American robins (Turdus migratorius). 2022. https://doi.org/10.1016/j.yhbeh.2022.105278
Reference
- Matsumoto, Shin-ichi, et al. "Highly-sensitive simultaneous quantitation of glucosylsphingosine and galactosylsphingosine in human cerebrospinal fluid by liquid chromatography/tandem mass spectrometry." Journal of Pharmaceutical and Biomedical Analysis 217 (2022): 114852. https://doi.org/10.1016/j.jpba.2022.114852







