C-Terminal Sequencing Analysis Service
Resolve Protein and Peptide C-Termini with Confidence
Pinpoint and validate the C-terminal residue of your protein or peptide—detect truncation, verify modifications, and ensure batch consistency with high-resolution LC–MS/MS.
- Resolve C-termini unambiguously: Carboxypeptidase laddering + high-resolution MS/MS
- Catch micro-heterogeneity: Truncations, C-terminal Lys loss, amidation, esterification
- Map protease cleavage sites: Discovery-scale C-terminomics for neo-C-termini
- Biologics-ready: Antibody heavy/light chain C-termini, peptide therapeutics, engineered enzymes
Submit Your Request Now
×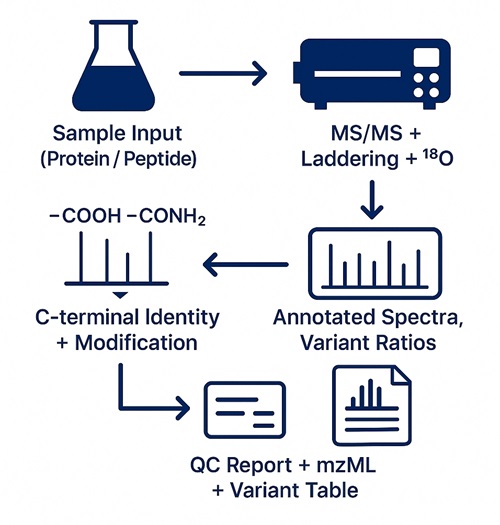
What You Receive:
- Raw MS files (.raw / .wiff) and converted mzML
- Annotated MS/MS spectra of C-terminal peptides (PDF or PNG)
- C-terminal variant tables with abundance, sequence, modification state
- Laddering summary (if applied), 18O confirmation (if used)
- FDR, localization score, and QC summary report
- Optional: neo-C-termini motif logo, cleavage site table
- What We Provide
- Advantages
- Technology Platform
- Sample Requirement
- Demo
- FAQs
What Is C-Terminal Sequencing
C-terminal sequencing refers to a collection of specialized proteomic techniques that enable direct identification of the final amino acid residue(s) in a protein or peptide, as well as characterization of any chemical modifications at the terminal carboxyl group. It addresses a blind spot often left unresolved by conventional bottom-up proteomics, which rarely capture the terminal peptide due to enzymatic cleavage limitations or poor ionization efficiency.
Common C-Terminal Challenges in Protein Characterization:
- Unrecognized truncation or clipping, introduced during expression, purification, or storage
- C-terminal lysine heterogeneity in monoclonal antibodies, impacting charge-based separation and stability
- Incorrect or incomplete chemical modifications, such as amidation or esterification in synthetic peptides and tags
- Neo-C-termini formation due to protease activity, degradation, or structural rearrangement
These C-terminal alterations can undermine protein function, batch comparability, and regulatory consistency, particularly in biologics and peptide-based therapeutics. Accurate terminal mapping is therefore essential for confident characterization and risk mitigation.
What You Achieve With C-Terminal Sequencing
Creative Proteomics's C-terminal sequencing workflows are designed to produce defensible, high-resolution data at the molecular endpoint. This enables you to:
- Verify the precise terminal amino acid, ensuring structural completeness and sequence accuracy
- Quantify minor variants such as clipped forms, amidated species, or des-Lys populations—down to ~1–2% abundance
- Differentiate chemical states of the C-terminus (–COOH vs –CONH₂, methyl/ethyl esters), which affect bioactivity and formulation
- Assess C-terminal integrity across batches, stress conditions, or expression systems for comparability and QA
- Support protease mapping and degradation studies by identifying neo-C-termini with high specificity
- Meet documentation and regulatory needs with annotated spectra, variant ratios, and QC-metric reporting
Each dataset is tailored to your study goals—whether verifying a purified biologic or exploring terminal heterogeneity across complex proteomes.
What Creative Proteomics Provides: Specialized C-Terminal Sequencing Services
C-Terminal Verification for Intact Proteins (e.g., mAbs, Enzymes)
Purpose: Confirm the final amino acid residue and detect truncation or heterogeneity.
Strategy:
- Carboxypeptidase laddering (CpY/CpB) for mass-based residue-by-residue confirmation
- Optional native MS or intact Orbitrap-MS for step-down mass profiling
- Detection and quantification of C-terminal Lys variants (Lys⁺ / Lys⁻)
Best for:
- Monoclonal antibodies, fusion proteins, enzymes
- Release testing, stability assessments, lot comparability
Peptide API C-Terminus Chemistry Confirmation
Purpose: Determine if the peptide is correctly amidated, esterified, or clipped.
Strategy:
- PRM/MRM or HCD-based LC–MS/MS targeting terminal fragments
- Optional derivatization workflows to distinguish –COOH vs –CONH₂
- Detection of Gly-extended precursors, capped variants, or incomplete synthesis products
Best for:
- Therapeutic peptides, modified peptide APIs, QC of synthetic batches
Discovery-Mode C-Terminomics for Protease Profiling & Degradation Mapping
Purpose: Identify all native and neo-C-terminal peptides in complex samples.
Strategy:
- Enrichment via COFRADIC- or C-TAILS–like diagonal fractionation
- Enzyme-nonspecific database search with FDR control & motif analysis
- Outputs include neo-C-terminal site lists, cleavage motif heatmaps, and quantitative shifts
Best for:
- Protease characterization, degradation pathway analysis, stress testing
- Lysates, tissue extracts, cell culture samples
Tag, Linker, and Fusion Protein C-Terminus Validation
Purpose: Confirm the presence and chemical integrity of engineered C-terminal regions.
Strategy:
- Laddering or targeted PRM to verify tag/linker presence
- Detection of proteolytic loss, esterification, or truncation during purification
- Compatibility with conjugates, payloads, or modification sites
Best for:
- Tagged proteins, engineered biologics, ADCs, biosensors
- Preclinical development and construct verification
Advantages of C-Terminal Sequencing Service
- Single-residue terminal localization ≥99% confidence, supported by ladder and MS/MS ion evidence
- Variant detection to ~1–2% relative abundance; reporting threshold configurable per study
- Modification-Specific Workflows: Distinguishes –COOH, –CONH₂, and esterified termini; LOQ ≤10 fmol
- Quant precision: PRM/MRM CV ≤10% (technical), XIC-based relative quant with retention-time locking
- FDR control ≤1% (PSM-level); stricter filters for terminal peptides; mass accuracy ≤3 ppm typical
- Low-input compatibility: from 5–10 µg peptide or ~20 µg purified protein (method-dependent)
- Orthogonal confirmation combining laddering + 18O + enrichment, reducing false localization
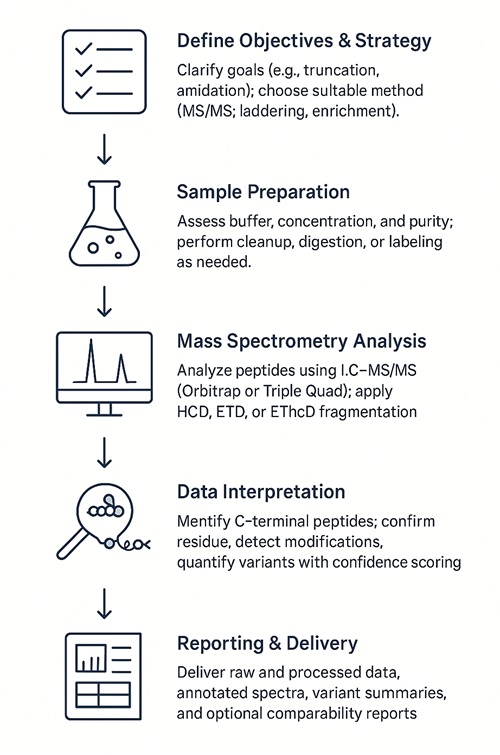
Workflow for C-Terminal Sequencing Analysis Service
1. Project Scoping
Define study objective (verification or discovery); identify expected C-terminal variants (e.g., truncation, amidation); select analysis strategy (MS/MS, laddering, enrichment).
2. Sample Evaluation & Preparation
Assess sample quality and buffer; perform cleanup, desalting, and optional reduction/alkylation; apply digestion or enrichment protocol based on project needs.
3. Mass Spectrometry Acquisition
Analyze samples via LC–MS/MS using Orbitrap or Triple Quad systems; select appropriate fragmentation mode (HCD, ETD, EThcD); acquire intact mass if laddering is used.
4. Data Processing & Validation
Perform terminal-aware database search; validate residue-level assignments; detect and quantify C-terminal variants; apply FDR control and localization scoring.
5. Reporting & Delivery
Provide raw data and processed files (mzML); deliver annotated spectra, variant tables, and QC summary; optional comparability and regulatory-ready reporting.
Technology Platform for C-Terminal Sequencing Analysis Service
C-terminal sequencing is typically performed using three core strategies: tandem mass spectrometry (MS/MS), carboxypeptidase digestion, and chemical cleavage. Among these, MS/MS is the preferred method due to its precision, speed, and broad applicability.
Tandem Mass Spectrometry (MS/MS) — Primary Method
We apply high-resolution LC–MS/MS and MALDI–TOF to identify C-terminal peptides and modifications directly from digested protein samples.
- Resolves terminal residues with ≥99% confidence
- Detects variants (e.g., truncation, amidation) down to ~1–2%
- Supports blocked or modified C-termini
- Compatible with 18O labeling and C-terminal enrichment
Carboxypeptidase Digestion
Enzymatic removal of C-terminal residues using CpY or CpB produces a mass ladder, enabling residue-by-residue confirmation via LC–MS or native MS.
- Ideal for intact protein verification (e.g., mAbs)
- Detects C-terminal Lys variants and minor truncations
- Often used as an orthogonal check
Chemical Cleavage
Cleaves near the C-terminus using harsh reagents, followed by MS analysis.
Now rarely used due to low specificity and sample compatibility limitations.
Platforms We Use
| Instrument | Use | Key Features |
|---|---|---|
| Orbitrap Exploris / Q Exactive | Peptide mapping | HCD/ETD; ≤3 ppm accuracy |
| TSQ Altis (Triple Quad) | Variant quantification | PRM/MRM; LOQ ≤10 fmol |
| MALDI–TOF | Rapid screening | High-throughput, intact mass check |
| CpY/CpB + LC–MS | Terminal confirmation | Ladder-based residue validation |

Agilent 6495C Triple Quadrupole (Figure from Agilent)

TSQ Altis Triple Quadrupole MS (Figure from Thermo Scientific)

SCIEX Triple Quad™ 6500+ (Figure from Sciex)
Sample Requirements & Compatibility
| Sample Type | Minimum Input | Buffer Compatibility | Notes / Recommendations |
|---|---|---|---|
| Purified Proteins (e.g., mAbs, enzymes) | ≥ 20–50 µg | Volatile buffers preferred (e.g., ammonium acetate); salt ≤ 50 mM | Avoid high glycerol/SDS; indicate disulfide status and post-translational modifications |
| Peptide APIs / Synthetic Peptides | ≥ 5–10 µg | Water, 0.1% FA, or mild buffers; avoid primary amines | Specify C-terminal modifications (e.g., amidation); glycine-extended forms can be assessed |
| Cell Lysates / Complex Matrices | ≥ 100–300 µg total protein | Urea ≤ 1 M; SDS < 0.03% after cleanup | Provide lysis buffer details; reduction/alkylation may be required |
| Fusion Proteins / Conjugates | ≥ 20–40 µg | Avoid PEGylated or strongly buffered formulations | State tag/linker sequence; indicate expected modifications or truncation risks |
| Formulated Samples / Stability Study Material | Case-by-case evaluation | Submit formulation composition if possible | We can assess compatibility and advise desalting/preprocessing if needed |
Demo Results
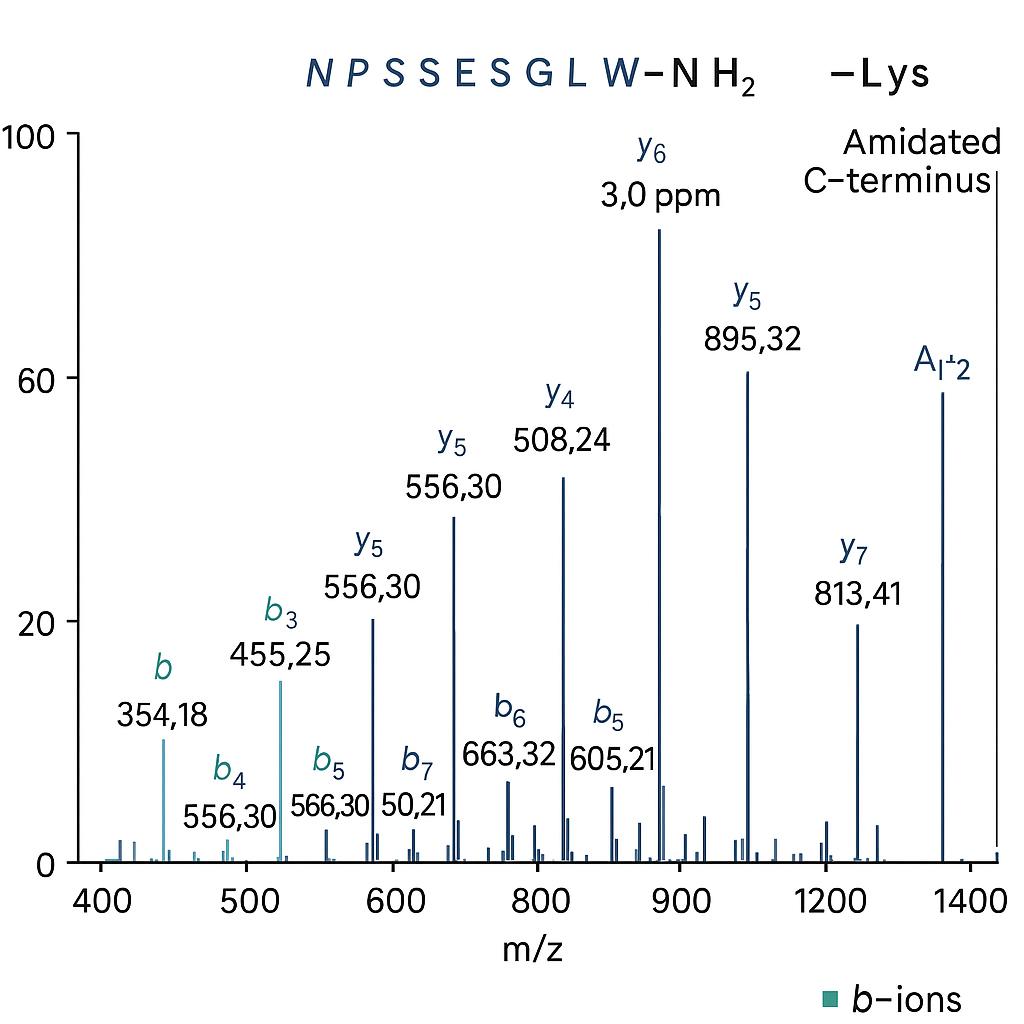
Annotated MS/MS spectrum confirming the C-terminal sequence and amidation via b/y ion series.
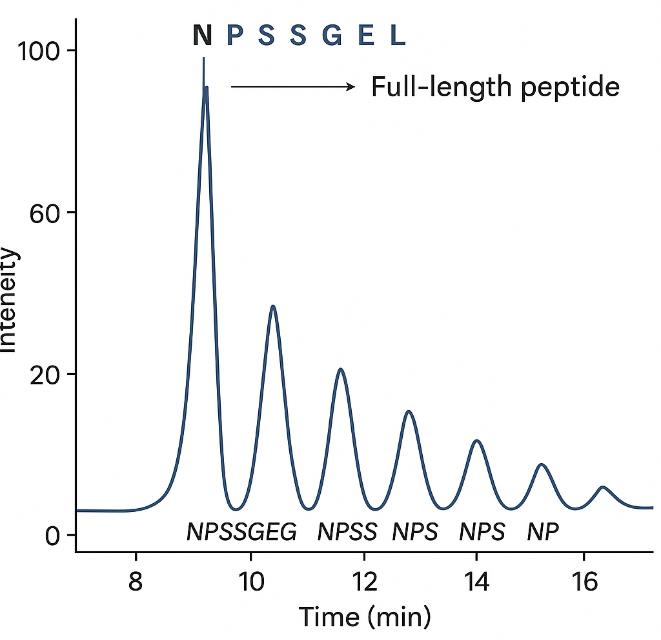
LC–MS profile showing sequential C-terminal residue loss after carboxypeptidase digestion.
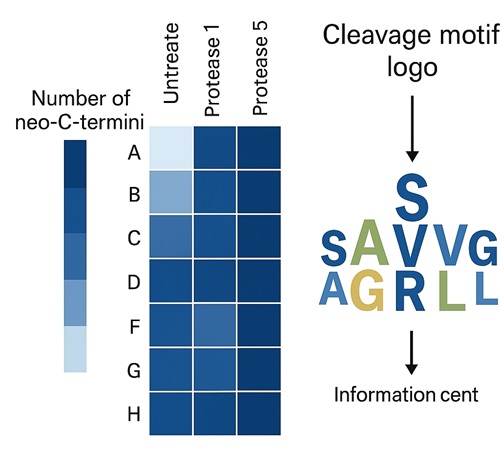
Neo-C-terminal mapping across protease treatments with sequence motif logo of cleavage sites.
Use Cases for C-Terminal Sequencing Analysis
Antibody C-Terminal Heterogeneity Assessment
Detect and quantify C-terminal Lys⁺/Lys⁻ variants in monoclonal antibodies. Confirm full-length heavy and light chains, identify stress- or process-induced C-terminal clipping, and support charge variant characterization during release testing or comparability studies.
Peptide API Identity and Modification Verification
Validate the C-terminal sequence and chemical state (e.g., –COOH, –CONH₂, esterified) of synthetic peptides used in therapeutic or diagnostic applications. Confirm amidation efficiency, detect glycine-extended or truncated variants, and ensure structural integrity across lots.
Fusion Protein Tag Confirmation and Linker Integrity
Confirm presence, sequence, and attachment site of C-terminal tags (e.g., His-tag, HA-tag, linker-payload) in recombinant proteins. Identify unintended truncations or cleavage events during expression, purification, or storage.
Protease Specificity Profiling and Neo-C-Termini Mapping
Map C-terminal cleavage sites generated by native or engineered proteases. Characterize proteolytic motifs and degradation hotspots using discovery-mode C-terminomics to study protease activity, drug-induced cleavage, or stability liabilities.
Bioprocess Monitoring and Stress Testing
Track C-terminal degradation or truncation under thermal, oxidative, or pH stress in biologics and recombinant proteins. Use quantitative C-terminal variant profiling to support formulation development, forced degradation studies, and stability-indicating assays.
FAQs of C-Terminal Sequencing
How do you confirm that a peptide represents the true C-terminus, not an internal fragment?
We require terminal-diagnostic ion series (b/y or c/z) in MS/MS spectra, apply localization scoring, and validate with orthogonal methods like ^18O labeling or carboxypeptidase laddering. Only calls meeting FDR, replicate, and spectral quality thresholds are reported.
Can you differentiate between free carboxyl, amidated, and esterified C-termini?
Yes. We use modification-aware workflows with derivatization-compatible MS and targeted transitions to clearly distinguish –COO⁻, –CONH₂, and esterified termini. Variant ratios can be provided when needed.
What is the sensitivity for detecting minor C-terminal variants or clipped forms?
We routinely quantify low-abundance variants (~1–2%) using PRM/MRM or XIC-based methods with strict RT locking, ion ratio filters, and QC acceptance gates to avoid overcalling noise.
Is this workflow applicable to antibodies, fusion proteins, or engineered constructs?
Absolutely. We verify C-terminal Lys⁺/Lys⁻ status in mAbs, confirm tag/linker integrity, and detect stress- or process-induced C-terminal degradation in fusion or conjugated proteins.
Can you map protease cleavage sites and neo-C-termini in complex samples?
Yes. Our C-terminomics enrichment workflows isolate native and neo-C-terminal peptides, enabling precise cleavage site mapping, sequence motif analysis, and condition-based comparisons.
How do you minimize false positives from sample handling artifacts?
We apply stabilizing chemistry, strict sample controls, and terminal-aware search filters. Any low-confidence or ambiguous sites are flagged with supporting evidence and confidence grading.
Are challenging targets like glycoproteins or disulfide-rich proteins supported?
Yes. We offer deglycosylation and reduction options, and use ETD or EThcD fragmentation to retain sequence coverage in labile or heavily modified C-terminal regions.
Can you identify blocked or conjugated C-termini?
Yes. We detect blocked termini (e.g., amidated, esterified, biotinylated, payload-conjugated) using selective chemistries and targeted MS readouts. Laddering can be added for confirmation.
What if my sample input is limited or matrix is complex?
We support low-input workflows and matrix-tolerant acquisition strategies with micro/nanoflow LC and optimized gradients while maintaining high terminal resolution and reproducibility.
Do you offer comparability analysis across lots or stress conditions?
Yes. We provide batch-aligned quantification with retention time normalization and internal references to track terminal variants across samples or production batches.
When is carboxypeptidase laddering recommended?
When residue-by-residue mass confirmation is needed—such as for verifying mAb C-termini or validating ambiguous MS/MS calls. It also serves as orthogonal evidence in regulatory settings.
How do you handle nearby PTMs that may interfere with C-terminal localization?
We use PTM-aware search strategies and fragmentation modes that retain labile sites. Terminal scoring accounts for PTM-induced ion shifts and spectrum complexity.
Which quantification approach do you recommend for confident reporting?
PRM/MRM is preferred for targeted, known variants. XIC is ideal for discovery-mode datasets, with targeted follow-up to validate critical or unexpected C-terminal forms.
Learn about other Q&A about proteomics technology.
Publications
Here are some publications in Proteomics research from our clients:

- Isolation of the mustard Napin protein Allergen Sin a 1 and characterisation of its antifungal activity. 2022. https://doi.org/10.1016/j.bbrep.2022.101208
- Functional Characterization of Propeptides in Plant Subtilases as Intramolecular Chaperones and Inhibitors of the Mature Protease. 2016. https://doi.org/10.1074/jbc.M116.744151
- Chemoproteomic identification of CO2-dependent lysine carboxylation in proteins. 2022. https://doi.org/10.1038/s41589-022-01043-1







