Introduction
Protein sequencing — the determination of the precise order of amino acids in a protein — is a foundational technique in molecular biology, biochemistry, and biopharmaceutical development. The amino acid sequence, also referred to as the primary structure, determines how a protein folds into its three-dimensional conformation, how it interacts with other molecules, and what biological functions it performs. From the earliest methods based on chemical degradation that could sequence a few residues per day, to modern mass spectrometry platforms capable of sequencing thousands of proteins in a single experiment, the field of protein sequencing has undergone a dramatic transformation over the past seven decades. This guide provides a technical overview of protein sequencing methods, a practical decision framework for selecting the appropriate approach based on research objectives and sample characteristics, and discusses key applications in drug development, structural biology, and proteomics. Each section is designed to help researchers understand not just the available methods, but how to choose between them and how to interpret the results.
The Significance of Protein Sequencing
Protein sequencing serves as the bridge between genomic information and functional biology. The genetic code stored in DNA is transcribed into messenger RNA and translated into proteins, but the sequence of the final protein product cannot always be predicted from the gene sequence alone. Alternative splicing, RNA editing, post-translational processing, and protein maturation events can produce protein sequences that differ substantially from the translation of the open reading frame. Direct protein sequencing provides the definitive experimental evidence of the actual amino acid sequence. The six key dimensions of its significance are described below.
In drug development, knowledge of the target protein sequence enables rational design of small molecule inhibitors that fit precisely into binding pockets, therapeutic antibodies that recognize specific epitopes, and peptide-based drugs that mimic natural ligands. The success of targeted cancer therapies such as kinase inhibitors and checkpoint inhibitor antibodies depends on accurate knowledge of the target protein sequence. For biopharmaceutical products, sequence confirmation is a regulatory requirement for batch release and comparability studies. The FDA and EMA require that the primary sequence of any recombinant therapeutic protein be confirmed by direct analytical methods, typically a combination of mass spectrometry-based peptide mapping and Edman degradation for N-terminal sequencing. Any sequence variant, truncation, or misincorporation detected during this analysis can trigger a full investigation into the manufacturing process.
In structural biology, the amino acid sequence is the starting point for X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy structure determination. Without an accurate sequence, electron density maps cannot be interpreted, and the three-dimensional structure cannot be solved.
In proteomics research, protein sequencing enables the identification of proteins in complex biological mixtures, the characterization of post-translational modifications that regulate protein function, and the quantification of protein expression changes between healthy and diseased states. These applications have driven the development of increasingly sensitive, high-throughput sequencing platforms that can analyze proteins from ever-smaller sample amounts.
The role of protein sequencing in biomedical research is expanding rapidly. Identifying protein variants, splice isoforms, and mutation-derived neoantigens requires sequencing methods that can detect single amino acid changes with high confidence. Mass spectrometry-based approaches coupled with genomic data are increasingly used in research settings, enabling the identification of tumor-specific neoantigens for preclinical immunotherapy studies. In addition, protein sequencing contributes to the characterization of genetic variants caused by protein-truncating mutations in research models, supports biomarker discovery studies for early disease detection, and enables monitoring of therapeutic protein levels in biological samples from preclinical studies.
Protein Sequencing Methods: From Classical to Cutting-Edge
Three principal approaches to protein sequencing are in current use, each with distinct capabilities, limitations, and optimal application domains. The choice between them depends on the sample characteristics, the research question, the available instrumentation, and the required throughput.
Edman Degradation
Figure 1: Protein Sequencing Workflow Overview
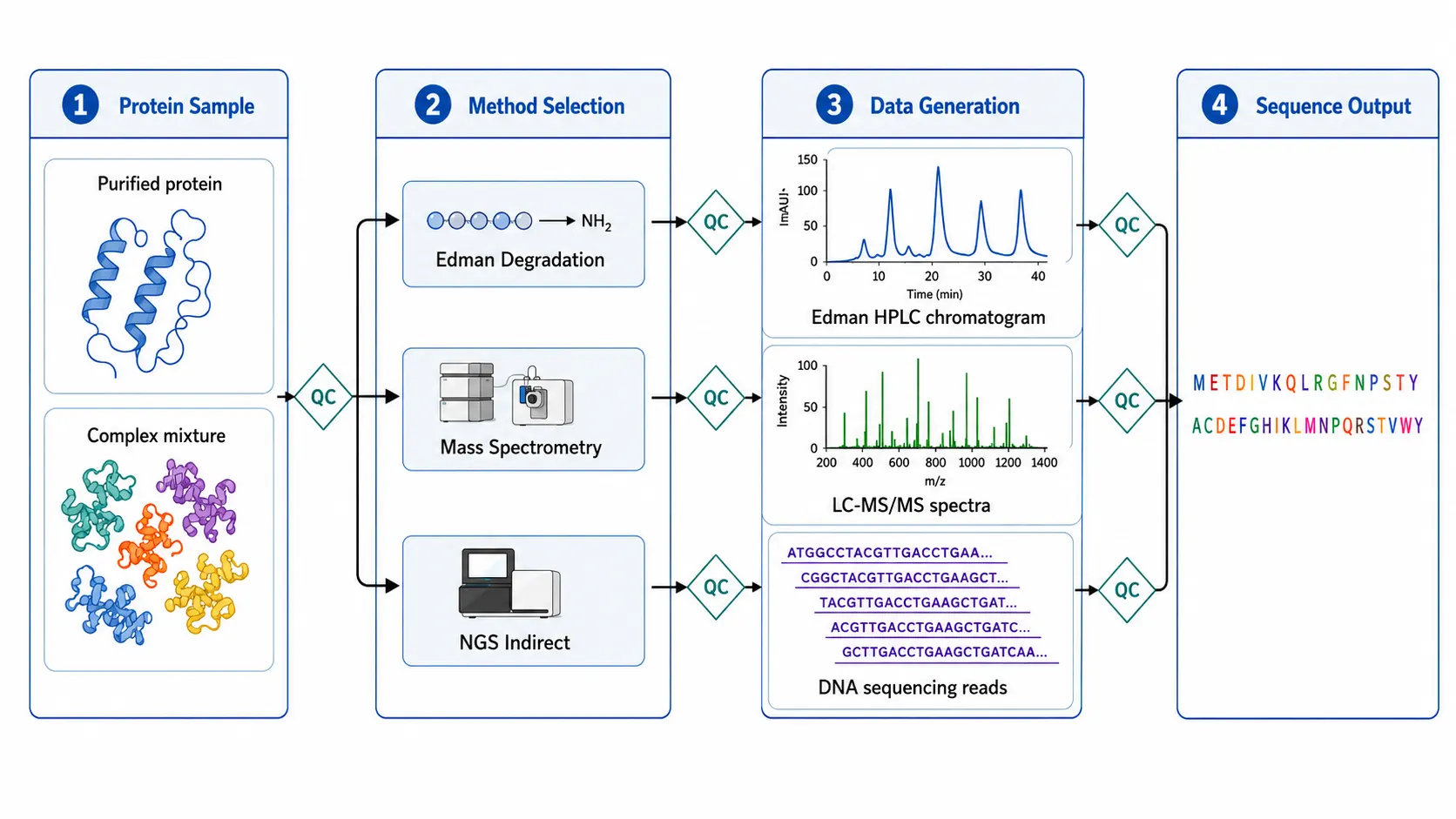
Figure 2: Edman Degradation Cycle Diagram
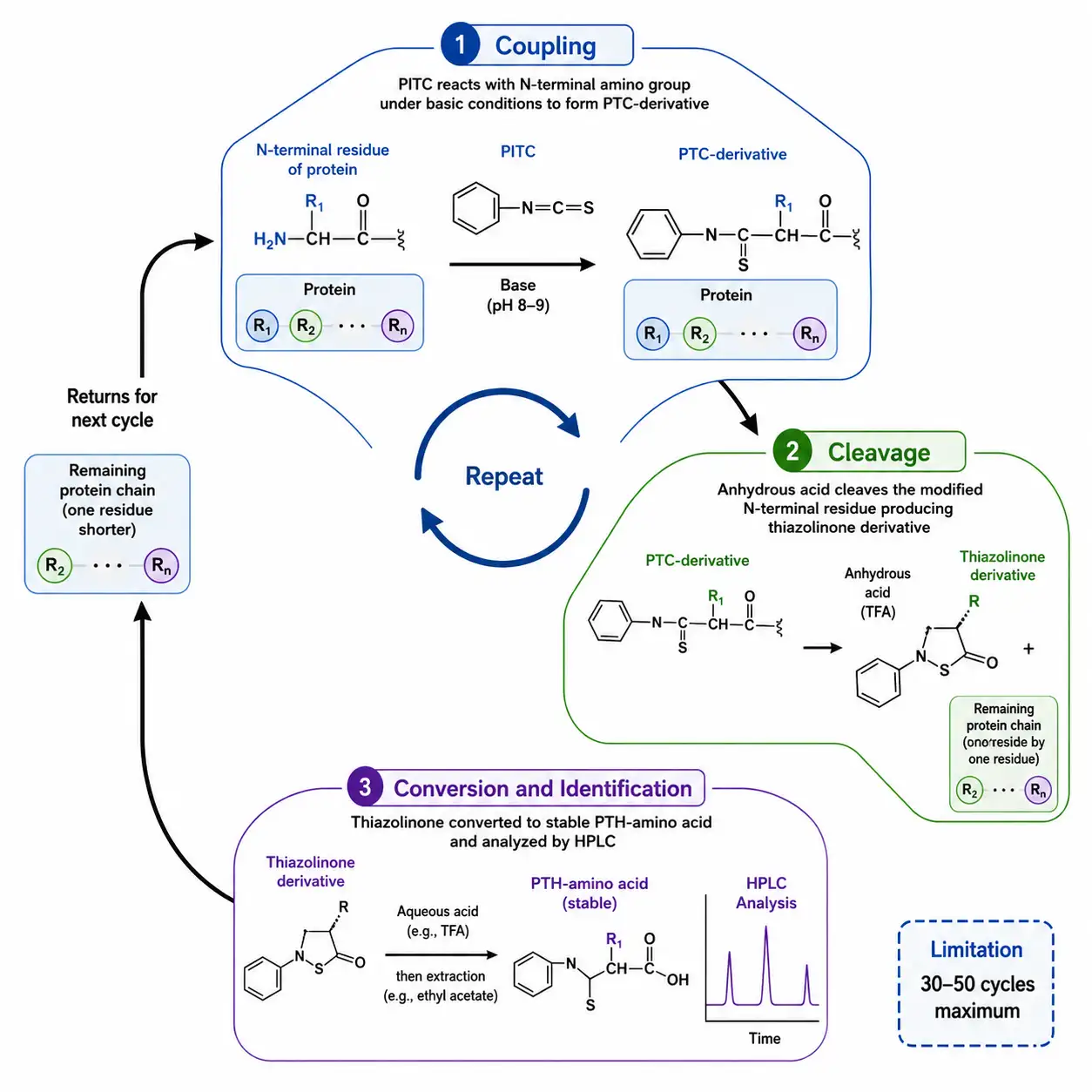
Edman degradation, developed by Pehr Edman in 1950, was the first practical method for determining the amino acid sequence of a protein. The method relies on a cyclic chemical reaction that sequentially removes and identifies the N-terminal amino acid. In each cycle, phenylisothiocyanate reacts with the free N-terminal amino group under mildly basic conditions to form a phenylthiocarbamyl derivative. Treatment with anhydrous acid cleaves the modified N-terminal residue, producing a thiazolinone derivative, while the remaining protein chain is left intact for the next cycle. The thiazolinone is converted to a more stable phenylthiohydantoin derivative and identified by high-performance liquid chromatography through comparison with PTH-amino acid standards.
Edman degradation offers several advantages. It provides direct, unambiguous sequence information without requiring a reference database or prior knowledge of the protein. Each amino acid is identified sequentially, and the result is a direct readout of the N-terminal sequence. The method is highly reliable for the first 20 to 30 residues and can, with optimized chemistry and instrumentation, extend to 50 or more residues from a few hundred picomoles of purified protein.
The limitations of Edman degradation are significant for modern applications. The method requires highly purified protein — contaminants produce mixed signals that cannot be deconvoluted. The sequencing yield decreases with each cycle due to incomplete coupling, cleavage, or extraction, and the cumulative loss limits the practical read length to approximately 30 to 50 residues. Edman degradation cannot sequence proteins with blocked N-termini, such as those modified by acetylation, pyroglutamate formation, or myristoylation. It provides no information about the C-terminal sequence or about internal regions unless the protein has been fragmented. And it is incompatible with post-translational modifications that are labile under the sequencing conditions.
Despite these limitations, Edman degradation remains valuable for specific applications. N-terminal sequence confirmation of recombinant proteins is a regulatory expectation for biopharmaceutical characterization, providing direct evidence that the expressed protein has the correct N-terminal processing. Sequencing of short peptides, verification of protein expression constructs, and quality control of synthetic peptides are routine applications where Edman degradation remains competitive with mass spectrometry. For proteins with blocked N-termini, enzymatic or chemical deblocking methods can remove certain modifications to enable Edman sequencing. Creative Proteomics provides an N-terminal Edman Degradation Service for applications requiring direct N-terminal sequence determination.
Mass Spectrometry-Based Protein Sequencing
Mass spectrometry has become the dominant technology for protein sequencing, offering unparalleled sensitivity, speed, and throughput. Modern mass spectrometry-based protein sequencing follows a well-established workflow: the protein is enzymatically digested into peptides, the peptides are separated by liquid chromatography, ionized by electrospray, and analyzed in the mass spectrometer. The mass spectrometer measures the mass-to-charge ratio of intact peptides in the first stage (MS1), selects individual peptides for fragmentation, and records the fragment ion masses in the second stage (MS/MS or MS2). The fragment ion masses are then used to determine the peptide sequence.
Figure 3: CID vs HCD vs ETD Fragmentation Comparison
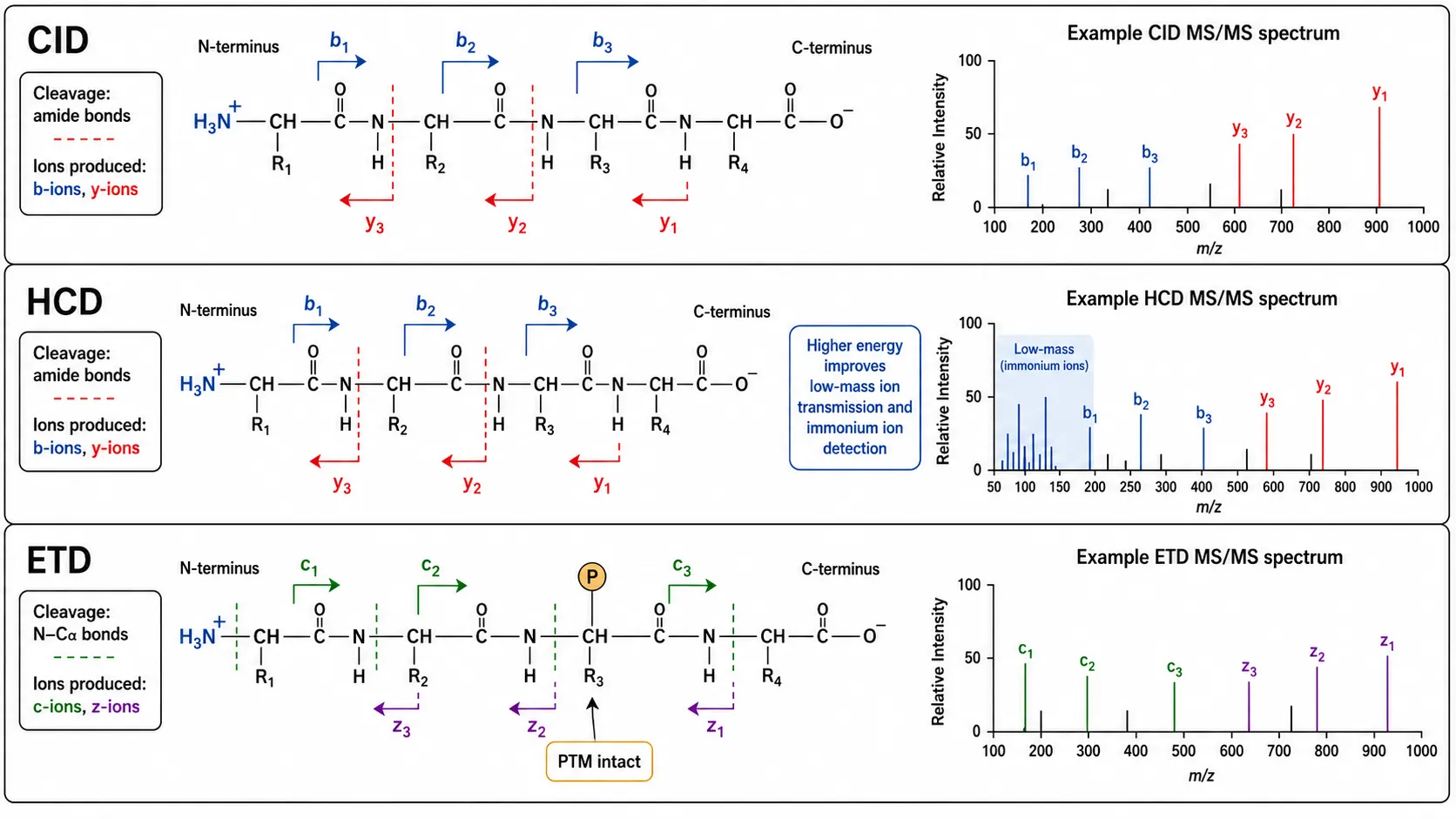
The standard fragmentation method in proteomics is collision-induced dissociation, in which selected peptide ions are accelerated and allowed to collide with inert gas molecules. CID primarily cleaves the amide bond along the peptide backbone, producing b-ions (fragment containing the N-terminus) and y-ions (fragment containing the C-terminus). The mass difference between consecutive fragment ions in a series corresponds to the mass of an amino acid residue, allowing the sequence to be read. The b-ion series provides sequence information from the N-terminus, while the y-ion series provides complementary information from the C-terminus. Together, they provide redundant sequence coverage that increases identification confidence.
Higher-energy collisional dissociation, available on Orbitrap mass spectrometers, uses a higher-energy collision cell to achieve more efficient fragmentation of larger peptides and to improve the detection of immonium ions that are diagnostic for specific amino acids. HCD also provides better transmission of low-mass fragment ions, enabling detection of phosphate neutral loss and other diagnostic fragments that aid PTM identification.
Electron transfer dissociation, available on ion trap and Orbitrap platforms, fragments peptides through a fundamentally different mechanism. ETD transfers an electron from a radical anion to the peptide cation, causing fragmentation along the N-C-alpha bond rather than the amide bond. This produces c-ions and z-ions that retain labile post-translational modifications such as phosphorylation and glycosylation, which would be lost during CID or HCD fragmentation. ETD is essential for sequencing peptides with labile modifications and for improving sequence coverage of highly charged peptides.
The choice of fragmentation method has a direct impact on sequencing success. CID and HCD are optimal for unmodified peptides and produce simpler spectra that are easier to interpret. ETD is preferred for phosphorylated, glycosylated, and other modified peptides where modification retention is critical. Hybrid approaches using both CID and ETD in the same experiment provide the most comprehensive sequence coverage. A practical strategy for comprehensive coverage is to acquire CID, HCD, and ETD spectra in the same LC-MS/MS experiment, a technique known as decision tree-based fragmentation. In this approach, the mass spectrometer automatically selects the optimal fragmentation method for each peptide precursor based on its charge state and mass. For peptides with a charge state of two or lower, CID or HCD is selected because these methods provide complete b-ion and y-ion series for short, low-charge peptides. For peptides with a charge state of three or higher, ETD is selected because these longer, more highly charged peptides fragment efficiently by electron transfer and retain labile modifications. The combined data set provides complementary sequence information that maximizes both sequence coverage and modification retention. Creative Proteomics De Novo Sequencing Service combines multiple fragmentation strategies with advanced bioinformatics for complete protein sequence determination.
NGS-Based Indirect Sequencing
Next-generation sequencing provides an indirect approach to protein sequencing by determining the sequence of the messenger RNA that encodes the protein. The mRNA is extracted from the cell or tissue of interest, converted to complementary DNA, and sequenced using high-throughput platforms. The protein sequence is inferred from the cDNA sequence using the genetic code.
This approach offers extraordinary throughput. A single NGS experiment on a platform such as Illumina NovaSeq can sequence tens of thousands of mRNAs simultaneously, generating billions of sequencing reads that provide sequence information for thousands of proteins in parallel. The depth of coverage allows detection of low-abundance transcripts and splice variants that would be missed by protein-level analysis alone. NGS is indispensable for transcriptome-wide studies, for discovering alternatively spliced isoforms that produce protein variants with different functions, and for characterizing the protein repertoire of organisms whose genome has not been sequenced. The cost per base of NGS has dropped by more than 100,000-fold since the human genome project, making transcriptome sequencing accessible to individual laboratories.
However, NGS-based protein sequencing has fundamental limitations. It provides only indirect sequence information — the actual protein sequence may differ from the translation of the mRNA due to RNA editing, alternative translation initiation, proteolytic processing, or post-translational modification. NGS cannot detect the hundreds of chemically diverse modifications that occur after protein synthesis, and it cannot confirm the actual sequence of the mature functional protein.
A Practical Decision Framework for Method Selection
Figure 4: Protein Sequencing Decision Framework Flowchart
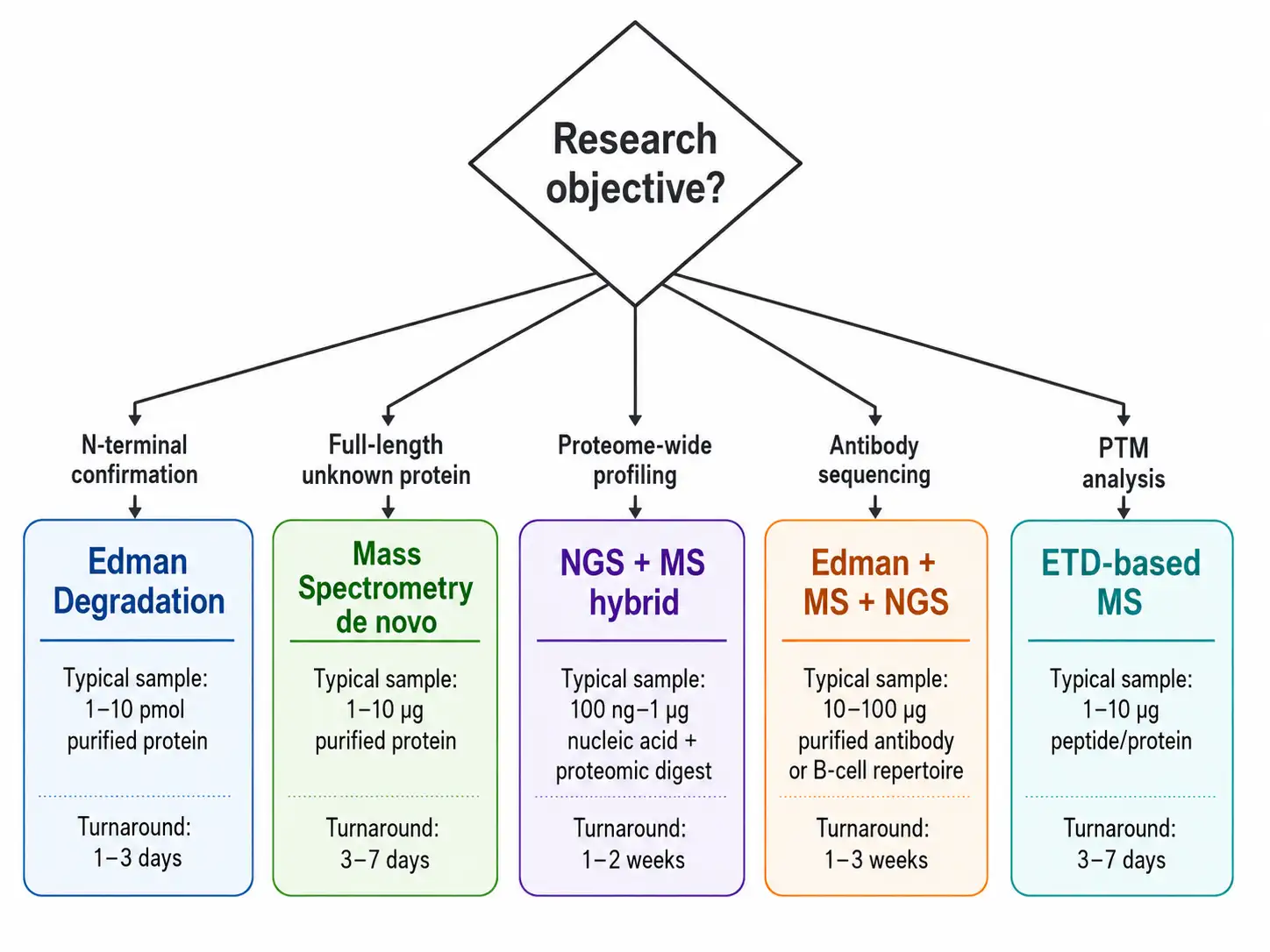
The choice between Edman degradation, mass spectrometry, and NGS-based approaches depends on the specific requirements of each application. The following framework guides method selection based on the research objective, sample characteristics, and required information.
For N-terminal sequence confirmation of recombinant proteins, Edman degradation is the method of choice. It provides direct, unambiguous N-terminal sequence information that satisfies regulatory requirements for identity testing in biopharmaceutical quality control. The method requires 10 to 100 picomoles of purified protein and provides sequence information for the first 10 to 30 residues. Mass spectrometry can also confirm the N-terminal sequence, but the interpretation is indirect and requires database searching or de novo sequencing, and blocked N-termini may go undetected without specific chemical treatment.
For full-length protein sequencing of unknown proteins, mass spectrometry-based de novo sequencing is the most practical approach. The protein is digested with multiple proteases, the resulting peptides are analyzed by LC-MS/MS with CID, HCD, and ETD fragmentation, and the sequences are assembled into the full-length protein using specialized bioinformatics tools. This approach can sequence proteins up to 100 kilodaltons or more from as little as 1 microgram of protein.
For comprehensive proteome-wide analysis, NGS of the transcriptome followed by mass spectrometry-based proteomics provides complementary information. NGS provides the sequence template, and mass spectrometry confirms the expression and modification state of individual proteins. This hybrid approach is the standard in modern proteomics.
For antibody sequencing, a combination of Edman degradation for light and heavy chain N-termini, mass spectrometry-based de novo sequencing for full-length sequence determination, and NGS of the hybridoma or B-cell mRNA for confirmation provides the most comprehensive and reliable sequence. The Edman data confirms the N-terminal sequences and establishes the exact N-terminal processing. The mass spectrometry data from multiple protease digests provides overlapping peptide coverage across the entire sequence, including the complementarity-determining regions that determine antigen binding specificity. The NGS data provides the cDNA sequences of the light and heavy chains, which can be used to confirm the protein-level sequences and to identify any discrepancies due to post-translational processing or sequencing errors. Creative Proteomics De Novo Antibody Sequencing Service integrates all three approaches for complete antibody sequence determination.
For PTM analysis, mass spectrometry with ETD fragmentation is the only practical method. The labile modifications are preserved during ETD fragmentation, enabling confident localization of phosphorylation, glycosylation, and other modifications to specific residues.
Fragmentation Methods and Their Impact on Sequencing
The fragmentation method used in mass spectrometry-based protein sequencing directly determines the quality and completeness of the sequence information obtained. Understanding the strengths and limitations of each method is essential for designing experiments that yield the required information.
Collision-induced dissociation produces predominantly b-ions and y-ions from the peptide backbone. CID spectra are relatively simple and easy to interpret, with the most abundant fragment ions providing clear sequence information. CID is effective for unmodified tryptic peptides between 7 and 25 residues in length. For longer peptides, the internal energy distribution becomes less uniform, and fragmentation becomes less predictable. For shorter peptides, there may be insufficient fragments for confident sequence assignment. CID tends to fragment preferentially at proline and adjacent to acidic residues, producing characteristic spectral patterns that experienced analysts can recognize.
Higher-energy collisional dissociation produces similar fragment ion types to CID but with higher efficiency and better low-mass ion transmission. HCD spectra contain more complete b-ion and y-ion series than CID spectra for most peptides, resulting in higher sequence coverage and more confident identifications. HCD is the default fragmentation method on Orbitrap platforms and is suitable for most routine protein sequencing applications.
Electron transfer dissociation produces c-ions and z-ions by a fundamentally different mechanism that does not depend on vibrational activation. The key advantage of ETD is that labile post-translational modifications — phosphorylation, glycosylation, acetylation, ubiquitination — remain attached to the peptide backbone during fragmentation, allowing the modified residue to be identified and localized. ETD is most effective for peptides with a charge state of three or higher, which means it performs best on longer, more highly charged peptides. For short, singly or doubly charged peptides, ETD efficiency is low, and CID or HCD is preferred.
The decision tree approach to fragmentation method selection automates the choice between CID, HCD, and ETD based on precursor charge state and mass, maximizing both sequence coverage and modification information in a single LC-MS/MS experiment.
Database Search, De Novo Sequencing, and Hybrid Strategies
Figure 5: Database Search vs De Novo Sequencing Workflow
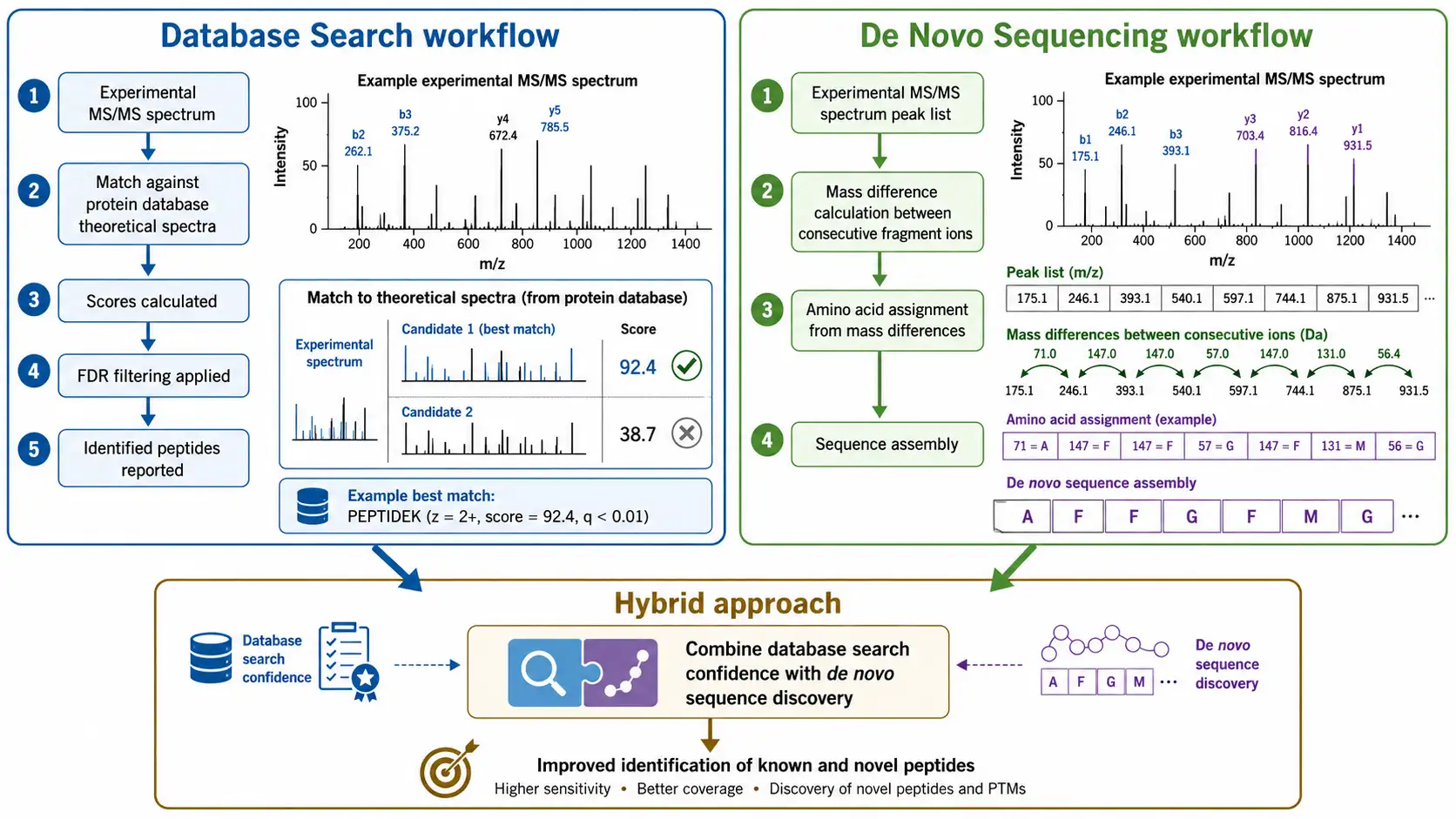
Mass spectrometry data can be interpreted by three fundamentally different strategies, each with distinct strengths and limitations.
Database searching compares experimental MS/MS spectra against theoretical spectra calculated from a protein sequence database. The search algorithm calculates a score for each database peptide based on how well the experimental and theoretical spectra match, and the highest-scoring match is reported as the identification. Database searching is fast, sensitive, and statistically well-characterized, with well-established false discovery rate estimation methods. The limitation is that it can only identify peptides present in the database — it cannot identify novel proteins, unexpected splice variants, or unanticipated modifications.
De novo sequencing determines the peptide sequence directly from the MS/MS spectrum without any database. The algorithm calculates the mass difference between consecutive fragment ions and matches each difference to the mass of an amino acid residue. De novo sequencing is essential for characterizing proteins from organisms without sequenced genomes, for identifying novel antibodies, and for detecting unexpected modifications. Modern de novo sequencing tools such as PEAKS, pNovo, and DeepNovo use machine learning to improve accuracy, achieving sequence accuracies above 90 percent for high-quality spectra. The main challenges are distinguishing leucine and isoleucine, which have identical masses, and handling incomplete fragment ion series that leave gaps in the sequence.
Hybrid strategies combine database searching and de novo sequencing to leverage the strengths of both approaches. In a typical hybrid workflow, database searching identifies peptides that match known sequences, and de novo sequencing identifies peptides that do not match any database entry. The unmatched peptides are then analyzed by homology searching against related species or by targeted de novo sequencing. Hybrid approaches provide the highest sequence coverage and are the standard strategy for comprehensive protein characterization.
Bottom-Up, Middle-Down, and Top-Down Approaches
Figure 6: Bottom-Up vs Middle-Down vs Top-Down Comparison
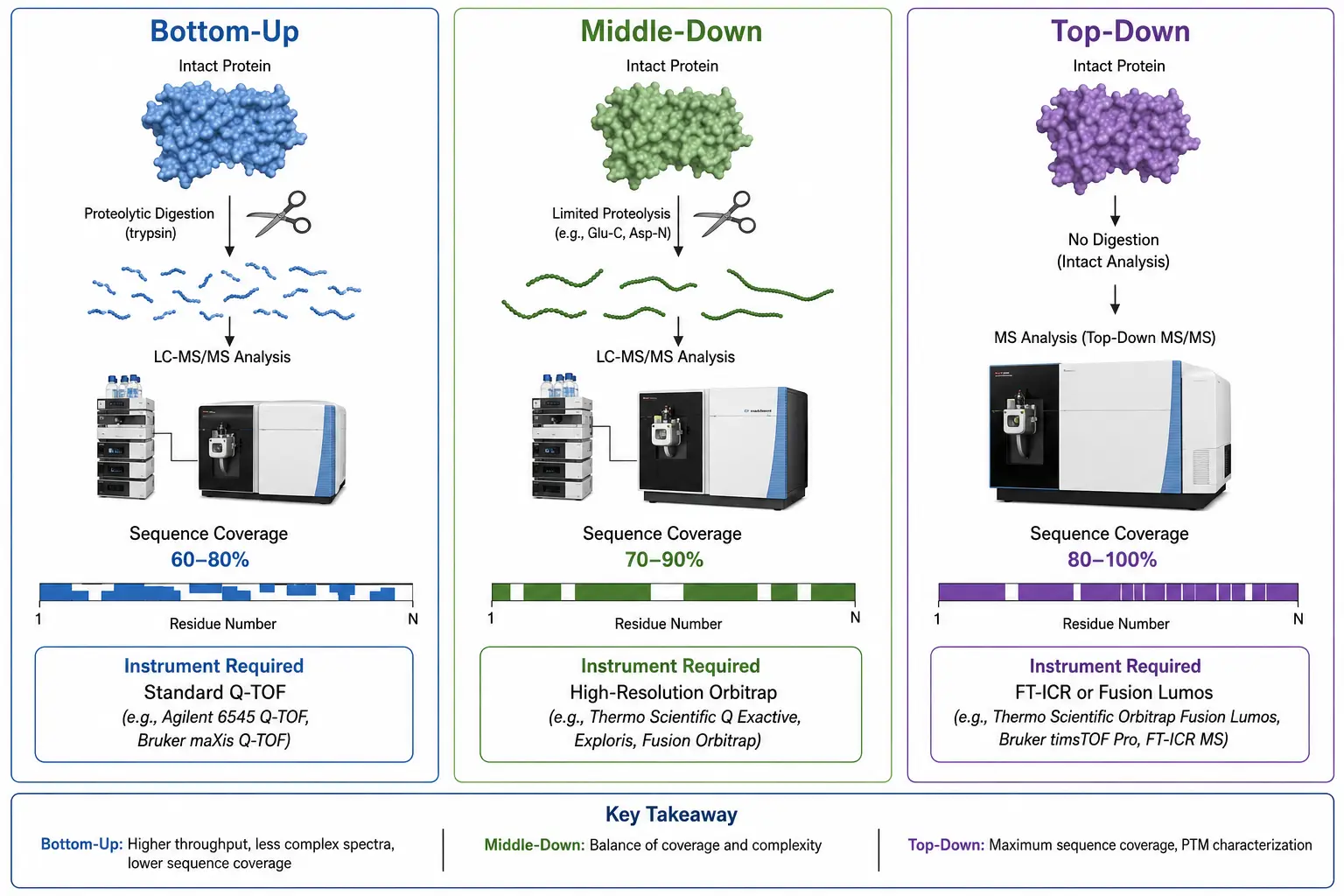
The scale at which protein sequencing is performed significantly affects the information obtained and the technical requirements of the experiment.
Bottom-up proteomics digests the protein into peptides averaging 7 to 25 residues before mass spectrometry analysis. This is by far the most common approach, used in over 90 percent of published proteomics studies, because tryptic peptides are well-behaved in reverse-phase chromatography, produce good CID spectra with complete b-ion and y-ion series, and can be identified with high confidence by database searching. The typical workflow uses trypsin as the primary protease, which cleaves after lysine and arginine residues, producing peptides with a basic C-terminal residue that ionizes efficiently in positive ion mode. The peptides are separated by reverse-phase liquid chromatography over a 60 to 120 minute gradient, ionized by electrospray, and analyzed by data-dependent acquisition where the most abundant peptide ions in each MS1 scan are selected for MS/MS fragmentation. A typical 60-minute gradient can analyze several thousand peptides, corresponding to several hundred to a few thousand protein groups. The limitation of bottom-up is that the connection between individual peptides is lost during digestion. The original protein sequence must be reconstructed computationally from the identified peptides, and this reconstruction is ambiguous when multiple protein isoforms share common peptides.
Middle-down proteomics uses less specific proteases or limited digestion conditions to produce larger peptides of 30 to 60 residues. Proteases such as Glu-C, Asp-N, or Lys-C, used alone or in combination with trypsin, produce longer peptides that retain more sequence context and improve the discrimination between protein isoforms. The larger peptides also provide better coverage of modification-rich regions because multiple modification sites may be present on a single peptide, allowing their co-occurrence to be detected. However, larger peptides are more challenging to fragment by CID or HCD, and they require higher-performance mass spectrometers with higher resolving power and mass accuracy. ETD is particularly useful for middle-down approaches because it fragments larger, highly charged peptides more efficiently than CID or HCD.
Top-down proteomics analyzes intact proteins without any digestion. The intact protein is introduced into the mass spectrometer, its mass is measured with high accuracy, and the protein is fragmented directly in the gas phase using electron capture dissociation or ETD to produce sequence information across the entire protein. Top-down sequencing provides the most complete sequence information possible from mass spectrometry, including the connectivity of post-translational modifications across the protein. It can distinguish closely related proteoforms that differ by a single modification, a single amino acid substitution, or a splice variation, providing information that is inaccessible to bottom-up approaches. The limitations are significant: top-down sequencing requires high-resolution mass spectrometers such as Orbitrap Fusion Lumos or FT-ICR instruments, the front-end separation is technically challenging because intact proteins are difficult to chromatograph, and the spectral complexity makes data interpretation more difficult.
For most routine applications, bottom-up proteomics provides the best balance of throughput, sensitivity, and information content. When isoform resolution, complete modification characterization, or the detection of unexpected proteoforms is required, middle-down or top-down approaches are used. Creative Proteomics Sequence Analysis Service provides comprehensive protein sequencing across all three scales.
Applications of Protein Sequencing
Applications of protein sequencing span the entire drug development pipeline and extend into basic research, diagnostics, and biotechnology.
In drug development, protein sequencing is used to identify and validate drug targets, characterize therapeutic proteins, and confirm the sequence of biopharmaceutical reference standards. For antibody-based therapeutics, full-length sequencing of the heavy and light chains is essential for establishing the intellectual property position and for demonstrating comparability between the innovator product and biosimilar candidates. De novo antibody sequencing from purified antibody samples is a well-established service that determines the complete primary structure, including the complementarity-determining regions that define antigen specificity. The sequenced antibody can then be expressed recombinantly for further development, even when the original hybridoma or B-cell clone is no longer available.
In structural biology, accurate protein sequencing is a prerequisite for interpreting diffraction data, NMR spectra, and cryo-electron microscopy reconstructions. Sequence errors at this stage can lead to incorrect structural models and wasted resources. Direct protein sequencing by mass spectrometry provides an independent verification of the sequence before committing to months or years of structure determination effort.
In proteomics research, protein sequencing enables large-scale identification and quantification of proteins from complex biological samples. Shotgun proteomics experiments using data-dependent acquisition routinely identify 3,000 to 6,000 proteins from tissue extracts, cell lysates, or biofluids in a single LC-MS/MS run. Data-independent acquisition methods such as SWATH-MS extend coverage to 5,000 to 10,000 proteins by systematically fragmenting all precursor ions in defined mass windows. Quantitative proteomics using stable isotope labeling techniques such as SILAC for cell culture experiments, TMT or iTRAQ for multiplexed comparison of up to 16 tissue samples, and label-free approaches for direct comparison of MS1 peak intensities between runs measure changes in protein abundance between experimental conditions with coefficients of variation below 20 percent.
Figure 7: Quantitative Proteomics Strategies Comparison Infographic
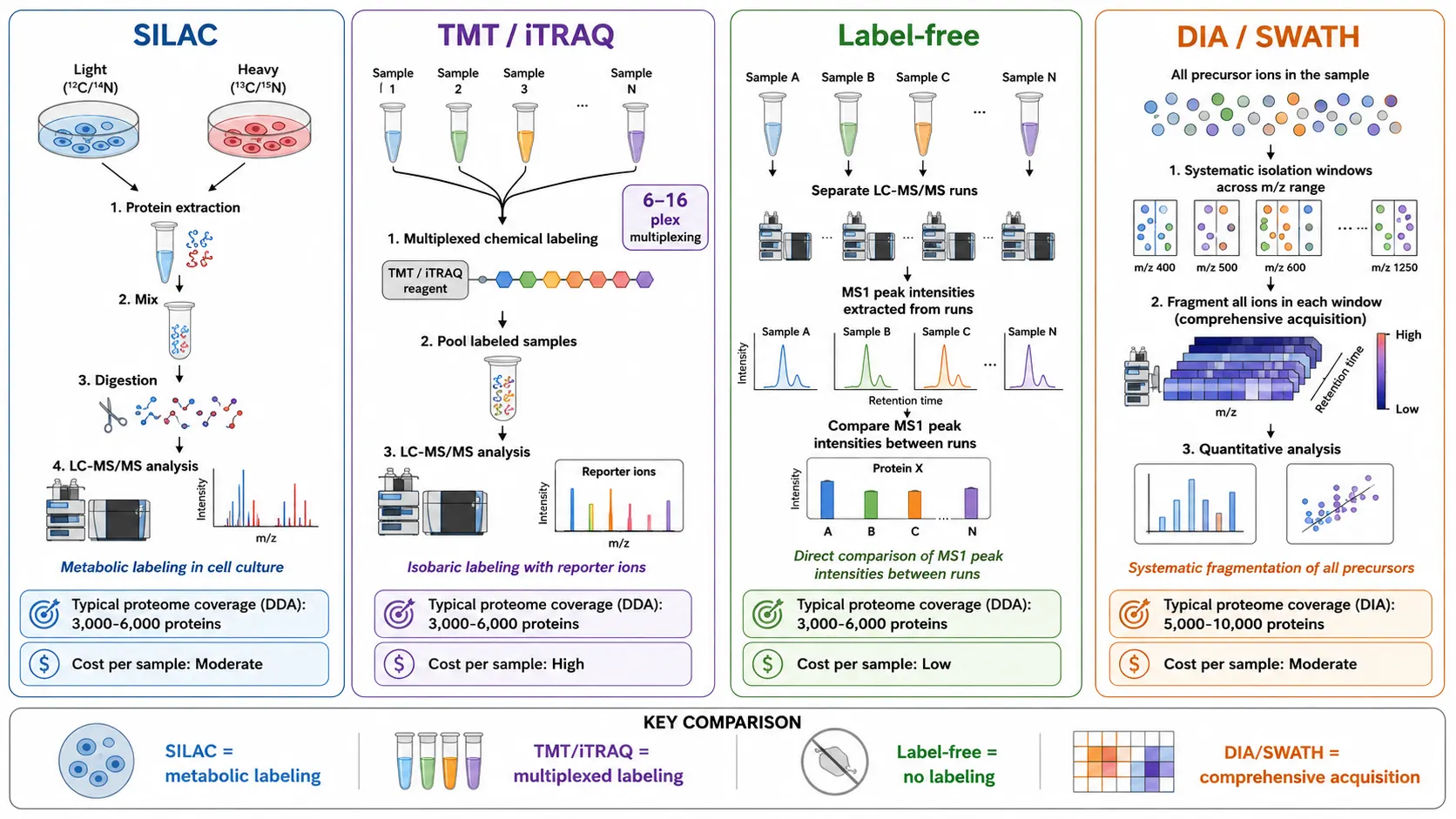
In biotechnology, protein sequencing is used to characterize recombinant proteins, confirm the identity of enzyme products, and troubleshoot fermentation or purification issues. Sequence analysis during process development detects unintended truncation, incorrect processing, or sequence variants that affect product quality. For cell line development, confirmation that the expressed protein matches the intended sequence is an essential quality checkpoint before advancing to manufacturing. In drug development research, protein sequencing identifies protein variants that affect drug metabolism and efficacy, supporting preclinical treatment strategy development. Mass spectrometry-based immunopeptidomics directly sequences peptides presented by major histocompatibility complex molecules, providing the molecular basis for preclinical cancer vaccine research. Peptide Mapping is a core technique for sequence confirmation and variant detection in biopharmaceutical development.
Challenges in Protein Sequencing and Practical Solutions
Sample preparation remains the most common source of difficulty in protein sequencing. Contaminants such as salts, detergents, and polymers suppress ionization in mass spectrometry and interfere with Edman chemistry. For mass spectrometry, the sample should be desalted by solid-phase extraction using C18 or polymeric reverse-phase cartridges, or by dialysis against a volatile buffer such as ammonium bicarbonate. Detergents should be avoided during sample preparation because they suppress electrospray ionization and produce intense background signals that mask analyte peaks. If detergents cannot be avoided, they must be removed by detergent removal spin columns or by phase transfer before mass spectrometry analysis. For Edman degradation, the protein must be purified to homogeneity — even 5 percent contamination from a second protein produces mixed signals that prevent reliable sequence interpretation. Buffer exchange into volatile buffers such as ammonium bicarbonate before analysis improves both mass spectrometry and Edman sequencing results by reducing the salt load that interferes with both techniques.
Data analysis complexity increases with the scale of the experiment. A typical bottom-up proteomics experiment analyzing a simple protein sample generates tens of thousands of MS/MS spectra that must be matched to peptide sequences. For a complex proteome sample, the number can exceed 100,000 spectra from a single LC-MS/MS run. False discovery rate control using target-decoy database searching is essential for reliable results, with a 1 percent FDR at the peptide and protein level being the standard for publication and regulatory submission. The target-decoy approach searches the experimental spectra against a combined database containing both forward (target) protein sequences and their reversed or shuffled (decoy) versions. The number of matches to decoy sequences estimates the false discovery rate, allowing the score threshold to be set at the desired FDR. For de novo sequencing, the confidence in each amino acid assignment should be reported alongside the sequence, and positions with low confidence should be verified by complementary fragmentation methods such as ETD to confirm labile modifications, or by analysis with alternative proteases to generate overlapping peptides that confirm the sequence.
Cost remains a practical consideration for large-scale proteomics studies. Bottom-up proteomics using label-free quantification is the most cost-effective approach for large sample sets, with per-sample costs of $200 to $500 for comprehensive analysis including sample preparation, instrument time, and data analysis. Multiplexed quantification using TMT or iTRAQ reduces the cost per sample by analyzing up to 16 samples simultaneously, bringing the cost per sample to $50 to $150 depending on the multiplexing level and the depth of analysis. Edman degradation is cost-effective for targeted N-terminal sequencing of individual proteins at $200 to $500 per sequence run of 10 to 20 residues, but becomes prohibitively expensive for whole-proteome studies.
Bioinformatics and Data Analysis Tools
The computational analysis of protein sequencing data is as important as the experimental methods themselves. Modern protein sequencing generates large volumes of data that require specialized bioinformatics tools for interpretation. The choice of software depends on the sequencing strategy, the mass spectrometer platform, and the specific research question. The scale of data generated by a single LC-MS/MS experiment — often tens of thousands of MS/MS spectra from a two-hour gradient — makes manual interpretation impractical, and automated computational analysis is essential for obtaining reliable results within a reasonable timeframe.
For database searching, the most widely used tools include Mascot, Sequest, Andromeda, and MS-GF+. These tools match experimental spectra against theoretical spectra calculated from protein sequence databases and provide statistical scoring of the matches. Mascot uses a probability-based scoring system reporting the expectation value for each match, while Sequest uses cross-correlation to compare experimental and theoretical spectra. Andromeda, integrated into the MaxQuant platform, provides fast searching with target-decoy FDR estimation. MS-GF+ uses a generating function approach for accurate statistical significance. The choice of search tool depends on the specific requirements of the experiment and the preferred data analysis platform, and many laboratories use multiple tools to maximize identification rates across diverse sample types.
For de novo sequencing, PEAKS is the most widely used commercial tool, combining de novo sequencing with database searching and PTM identification in a single platform. PEAKS achieves reported accuracies above 90 percent for high-quality spectra and provides confidence scores for each amino acid in the de novo sequence. pNovo is a free de novo sequencing tool developed for high-resolution HCD data using a graph-based approach that achieves competitive accuracy. DeepNovo and PointNovo use deep learning approaches to improve de novo sequencing accuracy, particularly for incomplete fragment ion series and noisy spectra where traditional graph-based algorithms struggle. These deep learning tools are trained on millions of spectra and can identify sequence patterns that are difficult to capture with rule-based approaches.
For hybrid approaches, tools such as PEAKS DB and Proteome Discoverer combine database searching and de novo sequencing in an integrated workflow that maximizes identification rates while detecting novel sequences. PEAKS DB first performs a database search to identify known peptides, then applies de novo sequencing to the remaining unmatched spectra, extending the identification coverage beyond the database. Machine learning algorithms trained on large spectral libraries are increasingly used to improve peptide identification, retention time prediction, and PTM localization. Deep learning tools such as DeepMass and Prosit predict peptide fragmentation patterns and retention times with high accuracy, enabling more sensitive database searching and providing confidence metrics for de novo sequencing results. In 2026, DiNovo was published in Nature Communications, achieving high-coverage de novo sequencing by using paired digestion with mirror proteases to generate complementary peptide sets. Bioinformatics for Proteomics provides integrated data analysis support for protein sequencing projects.
Emerging Technologies in Protein Sequencing
The field of protein sequencing is advancing rapidly, with several technologies that are extending capabilities beyond current mass spectrometry-based approaches. These developments promise to address current limitations in sensitivity, throughput, and the ability to analyze proteins at the single-molecule level.
Single-molecule protein sequencing using biological nanopores represents the most transformative emerging technology. Building on the success of nanopore DNA sequencing, engineered nanopores such as alpha-hemolysin, aerolysin, and MspA can distinguish all 20 standard amino acids and detect post-translational modifications including phosphorylation and glycosylation at the single-molecule level. Recent advances include electro-osmotic flow control to drive proteins through the pore without charge bias and unfoldase motor proteins such as ClpXP for translocation speed regulation at single-amino-acid resolution (Restrepo-Perez, Joo, & Dekker, 2018, Nature Nanotechnology, doi:10.1038/s41565-018-0236-6). Experts estimate fingerprint-based identification applications within 2 to 5 years and full de novo sequencing of unknown proteins within 5 to 10 years.
Deep learning continues to transform computational protein sequencing. PEAKS DeepNovo, PointNovo, and PowerNovo use convolutional neural networks and transformer architectures to achieve de novo sequencing accuracies exceeding 95 percent for high-resolution HCD spectra. These deep learning models are trained on millions of high-quality spectra and can identify sequence patterns that are difficult to capture with traditional graph-based algorithms. The integration of deep learning with hybrid database search and de novo sequencing workflows now represents the state of the art in computational proteomics, approaching the performance of database searching for well-characterized samples while maintaining the ability to detect novel sequences and unexpected modifications.
Frequently Asked Questions
What is the difference between Edman degradation and mass spectrometry?
Edman degradation sequentially removes N-terminal amino acids by chemical reaction, providing direct sequence readout for 30 to 50 residues. Mass spectrometry fragments peptides and measures fragment masses to determine sequences, enabling high-throughput analysis of complex mixtures.
How much protein is needed for sequencing?
Edman degradation requires 10 to 100 picomoles of purified protein. Mass spectrometry-based sequencing can work with as little as 1 microgram of protein or less.
Can sequencing distinguish leucine and isoleucine?
Standard mass spectrometry cannot distinguish these isomers. Edman degradation can distinguish them. Specialized MS approaches using high-energy fragmentation or ion mobility can partially resolve them.
What is de novo sequencing?
De novo sequencing determines the peptide sequence directly from MS/MS spectra without a database. Required for unknown proteins and antibodies.
How are post-translational modifications detected?
Mass spectrometry identifies modifications by characteristic mass shifts. ETD fragmentation preserves labile modifications for confident localization.
What is the accuracy of protein sequencing?
Database searching achieves greater than 99 percent accuracy. De novo sequencing accuracy ranges from 80 to 95 percent.
How long does protein sequencing take?
Edman: 2 to 4 hours for 20 residues. Mass spectrometry: 1 to 3 days including sample preparation and data analysis.
What is the difference between bottom-up and top-down?
Bottom-up digests proteins into peptides. Top-down analyzes intact proteins, preserving modification connectivity.
What is the maximum protein size for MS sequencing?
Bottom-up has no upper size limit. Top-down is practical up to approximately 50 kilodaltons on standard instruments.
How are antibodies sequenced?
By combining Edman degradation for N-termini, mass spectrometry de novo sequencing, and NGS for cDNA confirmation.
References
- Steen, H., and Mann, M. (2004). The ABC's (and XYZ's) of peptide sequencing. Nature Reviews Molecular Cell Biology, 5, 699-711. doi:10.1038/nrm1468
- Eng, J. K., McCormack, A. L., and Yates, J. R. (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry, 5(11), 976-989. doi:10.1016/1044-0305(94)80016-2
- Zhang, J., et al. (2012). PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Molecular and Cellular Proteomics, 11(4), M111.010587. doi:10.1074/mcp.M111.010587
- Tran, N. H., et al. (2019). Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods, 16, 63-66. doi:10.1038/s41592-018-0260-3
- Restrepo-Perez, L., Joo, C., and Dekker, C. (2018). Paving the way to single-molecule protein sequencing. Nature Nanotechnology, 13, 786-796. doi:10.1038/s41565-018-0236-6












