Meta Intent: A deep technical guide for scientists who need to preserve, interpret, and map native disulfide topology by LC-MS/MS without introducing analytical artifacts.
Disulfide bonds are often introduced as simple structural staples. That framing is too narrow. In real protein systems, a native disulfide pattern is a compact record of folding history, oxidative environment, and conformational integrity. It tells you more than whether cysteines were oxidized. It tells you whether the protein reached the right topology and whether that topology survived extraction, processing, and analysis.
That is why disulfide mapping matters in far more than antibody work. It matters in secreted enzymes, Fc-fusion proteins, cystine-knot peptides, recombinant glycoproteins, and many other disulfide-rich proteoforms. In all of these systems, the same basic risk appears. The disulfide arrangement present in the original sample is not always the same arrangement that reaches the mass spectrometer. If sample preparation creates reactive thiols, the topology can be rewritten before the measurement even begins.
This article focuses on that problem. It is not enough to find disulfide-linked species. The harder question is whether those linked species still represent the native state. That is why modern disulfide analysis has shifted away from bond detection alone and toward workflow design: how to preserve native topology, how to suppress scrambling, and how to choose fragmentation methods that produce real structural evidence rather than plausible-looking artifacts.
This article discusses research-use structural characterization workflows and does not address clinical diagnosis or therapeutic decision-making.
Why disulfide topology is structural evidence, not just a covalent feature
A disulfide bond is not merely a sulfur-sulfur connection. Inside a folded protein, it acts as a topological constraint. It limits conformational freedom, stabilizes local folds, and can determine whether distant sequence regions are allowed to approach each other at all. In proteins that contain several bridges, the full disulfide pattern behaves like a wiring diagram. One wrong pairing can shift the geometry of the whole molecule.
This is why intact mass alone is not enough. Two proteoforms can have the same overall mass and the same total sulfur content while carrying different connectivity. The instrument can confirm oxidation state without proving native linkage pattern. That distinction matters in structural characterization. A protein can appear consistent at the intact-mass level while still requiring topology-resolved characterization to confirm native disulfide connectivity.
The best way to think about disulfide mapping is to separate three questions:
- Was the protein oxidized?
- Was it oxidized into the native connectivity?
- Did the analytical workflow preserve that connectivity long enough to measure it?
Many workflows answer the first question. Fewer answer the second with confidence. The strongest workflows are designed around the third question first.
That is why disulfide analysis naturally overlaps with peptide mapping, top down proteomics, and broader structural characterization. The analytical target is not a bond in isolation. It is native topology.
The redox chemistry of cysteine
Cysteine is analytically difficult for the same reason it is biologically useful. Its sulfur can move between reduced and oxidized states under conditions that are common both in cells and in sample preparation. That gives cysteine strong structural and regulatory power. It also makes cysteine-rich proteins unusually vulnerable to workflow-induced artifacts.
In the reduced state, cysteine exists as a thiol, R-SH. In the oxidized disulfide state, two sulfur atoms form an R-S-S-R linkage. That bond is stable enough to reinforce tertiary structure, yet reactive enough to participate in exchange chemistry. Whether it stays intact depends on local environment, pH, solvent exposure, neighboring residues, and the presence of reactive thiolates.
The mechanistic turning point is thiolate formation. A neutral thiol is less reactive. A deprotonated thiolate is much more nucleophilic. Once a cysteine side chain enters that state, it can attack an existing disulfide and trigger thiol-disulfide exchange. In oxidative folding, that is useful. In analytical workflows, it is dangerous.
Local microenvironment matters here. A cysteine buried in a hydrophobic core behaves differently from one exposed on the protein surface. A nearby basic residue can shift effective reactivity. Solvent access can turn a normally quiet sulfur into a reactive one during denaturation or partial unfolding. This is why no universal preparation condition is equally safe for every protein. The same workflow can preserve one topology and distort another.
The chemistry also explains why disulfide bonds are not just "stability locks." In some proteins, they mainly reinforce a fold that has already formed. In others, they actively guide the folding pathway by narrowing the conformational search space. In multi-bridge proteins, wrong intermediates can appear first and then undergo isomerization before the native architecture emerges. That is exactly why biological systems do not leave disulfide formation to random oxidation.
Formation in the endoplasmic reticulum
Most secreted and membrane proteins form disulfides in the endoplasmic reticulum. The ER is not simply an oxidizing compartment. It is a controlled redox environment that promotes bond formation while also correcting mispaired intermediates.
Protein disulfide isomerase, or PDI, is central to this process. PDI can introduce a disulfide into a folding substrate, break a non-native linkage, and rearrange incorrect pairings until the protein reaches a more stable state. Ero1 helps reoxidize PDI so the catalytic cycle can continue. The important point is that biology does not treat disulfide formation as a single oxidation event. It treats it as oxidative folding under supervision.
That fact matters analytically. A native disulfide topology is often the endpoint of a curated pathway. Once that endpoint exists, it should be treated as fragile information. Harsh denaturation, permissive pH, and delayed thiol trapping can reopen the same exchange logic that the ER had to manage carefully in the first place.
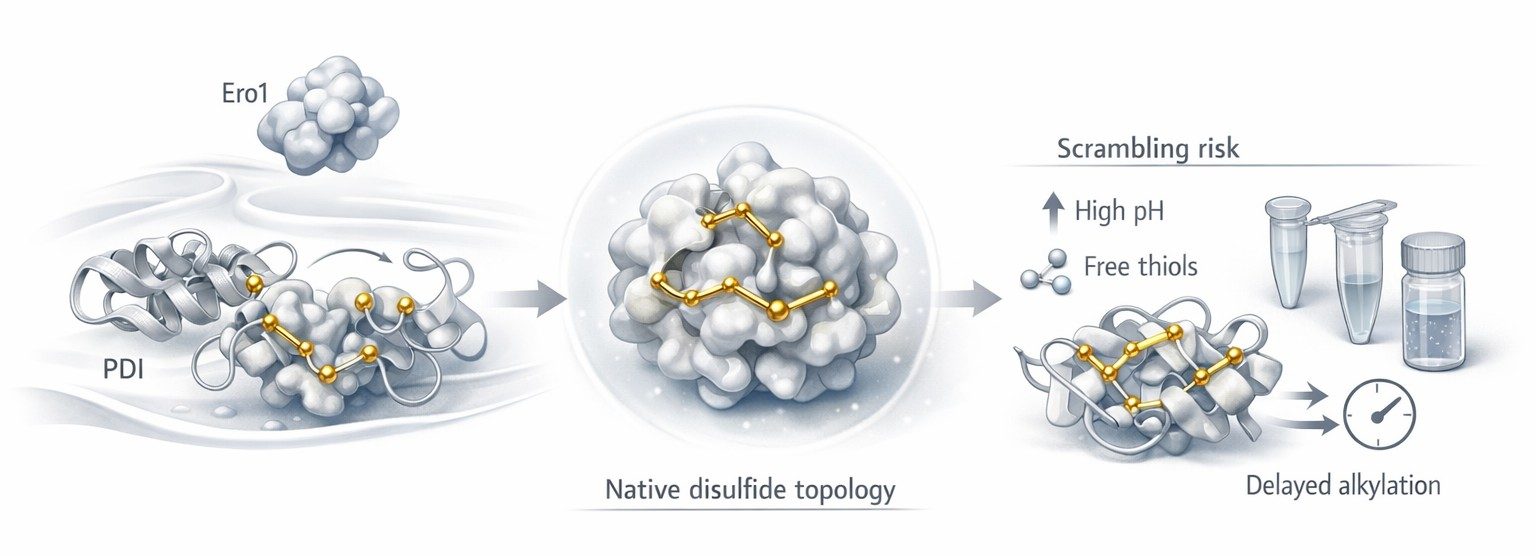 Figure 1: Oxidative folding and disulfide topology risk points
Figure 1: Oxidative folding and disulfide topology risk points
Figure 1 should appear here because this is the point where the biological and analytical stories meet. Up to now, the article has explained how native disulfides form. The figure should now help the reader see the transition from curated oxidative folding to post-extraction vulnerability. On the left, PDI and Ero1 support native topology formation. In the center, the folded protein represents the structurally correct reference state. On the right, sample preparation introduces the first major risk: the same sulfur chemistry that enabled folding can now generate non-native exchange.
The real analytical enemy: disulfide scrambling
Scrambling is often described too casually. It is not just "a possible artifact." It is the main reason a chemically plausible disulfide map can still be wrong.
In simple terms, scrambling means that a non-native disulfide arrangement was created during handling rather than being present in the original sample. The mechanism is straightforward. A free thiol becomes a reactive thiolate. That thiolate attacks an existing disulfide. One linkage breaks and another forms. The protein or peptide assembly still looks chemically credible. But the topology has changed.
This is what makes scrambling so dangerous. It rarely announces itself as catastrophic failure. More often, it appears as believable noise. Native linked species decrease. New linked species appear at lower abundance. Replicates become less consistent. The analyst may interpret those species as low-level heterogeneity, oxidation stress, or production-related variability. In reality, the sample may have been partially rewritten during preparation.
The risk becomes higher as topology becomes more complex. A protein with one disulfide bond offers fewer ways to go wrong than a protein with four, six, or eight bonds. Closely spaced cysteines, flexible loops, partially exposed bridges, and local unfolding all increase the number of topological alternatives that can emerge during handling. Once several reactive sulfur atoms are available in the same preparation window, chemistry starts exploring new pairings.
That is why scrambling has to be treated as the organizing principle of the workflow. If the workflow does not actively suppress thiol-disulfide exchange, the final map cannot automatically be trusted, no matter how elegant the LC-MS/MS data look.
Why pH is a topology-control variable
Most proteomics discussions treat pH as an enzyme-performance variable. In disulfide mapping, pH plays a different and more decisive role. It controls how easily cysteine enters the reactive thiolate state.
As pH rises, the fraction of deprotonated sulfur generally increases. That means more nucleophilic thiolate is present, and the system becomes more permissive to exchange. In ordinary peptide mapping, that may not be a major concern. In non-reduced disulfide analysis, it can determine whether the native bond pattern survives the preparation step.
This is why low-pH handling has become such an important control strategy. Under acidic conditions, usually around pH 2 to 3 for protective handling steps, thiol reactivity drops sharply. The sample is not frozen in an absolute sense, but it is much less able to undergo unwanted exchange. That is the real meaning of "freezing the disulfide state." The analyst is not just stopping one reaction. The analyst is shutting down the opportunity for a new topology to emerge.
This point is easy to miss because many teams inherit sample-prep workflows from standard peptide mapping. But a high-quality general digestion is not automatically a high-quality disulfide-preserving digestion. Those are different goals.
A practical rule helps:
- higher pH may support digestion convenience, but it also increases thiolate-driven risk
- lower pH suppresses exchange and protects native topology
- the best workflow balances enzyme access with structural preservation, rather than optimizing digestion alone
For proteins with multiple bridges, partially solvent-exposed cysteines, or redox-sensitive folds, that balance becomes the main experimental challenge.
Non-reducing digestion is not just standard digestion without DTT
This is a common mistake. Analysts often treat non-reducing digestion as routine digestion minus the reducing agent. That assumption causes trouble because removing DTT does not leave the rest of the logic unchanged. It changes the entire purpose of the preparation step.
Once reduction is removed, digestion must now do three things at the same time:
- generate peptides that can be analyzed by LC-MS
- preserve the native disulfide pattern
- avoid creating new free-thiol-mediated exchange pathways during unfolding and proteolysis
These goals do not align perfectly. Proteases often work best when the protein is accessible. Accessibility usually increases as the structure opens. But opening the structure can expose cysteines that were previously buried, destabilize certain local bridges, and lengthen the exchange window. Digestion time may improve peptide observability while making topology less trustworthy.
That is why non-reduced disulfide workflows often accept less than maximal sequence coverage in exchange for better topological fidelity. A complete digest that rewrites the sample is less useful than a more limited digest that preserves native linkages. In this context, digestion is not a routine technical step. It is a topology-preservation problem disguised as a protease step.
Careful protein sample preparation matters here for exactly that reason. The quality of a disulfide map depends on what happens before the peptides ever enter the LC column.
Alkylation is a kinetic race
Alkylation is often described as a housekeeping step. That language is misleading. In disulfide analysis, alkylation is a kinetic race against exchange, reoxidation, and side chemistry.
The goal seems simple. If free thiols are present, cap them before they can react. The difficulty lies in the word "before." Free thiols can re-form disulfides, attack native bridges, or remain partially accessible in unfolded or partially unfolded states. The alkylation reagent must therefore react quickly and selectively under conditions that do not create new ambiguity.
This is why reagent choice matters so much. The question is not merely whether a reagent can alkylate cysteine. The real question is how efficiently it closes the reactive thiol window in a topology-sensitive workflow.
IAM versus NEM
Iodoacetamide, or IAM, remains deeply familiar in proteomics. It fits well into many established data-analysis pipelines, and carbamidomethylation is a standard search modification. That makes IAM operationally convenient. But convenience should not be confused with universal suitability. IAM performance depends heavily on timing, pH control, and reaction management. If the sample remains exposed too long or the workflow is not tightly quenched, the free-thiol window may stay open longer than the analyst expects.
N-ethylmaleimide, or NEM, is often attractive when rapid thiol trapping is the priority. In many workflows, it caps accessible thiols quickly and helps suppress scrambling at an early stage. That benefit can be decisive when the sample is especially prone to exchange. But NEM also changes downstream interpretation. The mass shift is different, the search setup must reflect that, and reagent handling still needs to be controlled carefully.
The practical comparison is not "Which reagent is better in the abstract?" It is "Which reagent prevents unwanted thiol chemistry faster and more cleanly in this sample and this workflow?"
A useful decision rule is:
- choose IAM when the workflow is already tightly controlled and downstream compatibility is a major advantage
- choose NEM when fast thiol capture is the higher priority and scrambling suppression dominates the method design
- validate both in pilot work when the protein is disulfide-dense, conformationally fragile, or known to generate ambiguous linked species
In other words, alkylation should be judged by outcome, not by habit.
Over-alkylation is a hidden source of ambiguity
A poor alkylation step does not always fail by leaving thiols uncapped. It can also fail by modifying more chemistry than intended.
This is the problem of over-alkylation. In principle, alkylation should target sulfur. In practice, overly aggressive conditions can expand the reaction scope, complicate peptide populations, and make borderline assignments harder to defend. Lys-associated side chemistry is a common concern in IAM-heavy workflows, especially when reaction conditions drift away from what the sample actually needs.
Even low-level side reactions matter in disulfide work because the interpretation burden is already high. The analyst is not looking at ordinary peptides. The analyst is already dealing with linked species, structural constraints, and non-standard fragmentation patterns. Extra chemical heterogeneity pushes the data further away from clean assignment.
That is why alkylation conditions deserve the same design discipline as reduction and digestion conditions. Reagent excess, time, temperature, and quench behavior all affect whether the final map becomes clearer or more ambiguous.
Partial reduction is how complex topologies become solvable
Fully non-reduced mapping is powerful, but it reaches a limit in proteins that contain several closely spaced, nested, or intertwined disulfides. In those systems, a single digest can produce disulfide-linked assemblies that are real yet hard to interpret. The analytes become larger, the spectra become denser, and the topology can remain only partially resolved even when the chemistry was preserved correctly.
This is where partial reduction becomes especially useful.
Instead of breaking every disulfide at once, the analyst reduces only a controlled subset of bridges. That generates intermediate states. Some linked peptide assemblies fall apart. Others remain connected. By comparing these staged states, the analyst can infer which bonds were more accessible, which ones were more buried, and how the original architecture was organized.
That is the key conceptual advantage of partial reduction. It does not just simplify the sample. It reveals topology through sequence-controlled dismantling.
TCEP is often especially useful in this context because it supports precise, operationally clean reduction logic. Its value is not only that it reduces disulfides efficiently. Its value is that it can be used in a staged way. For complex proteins, that is a major difference.
A helpful distinction is this:
- full reduction asks what the sample looks like after all disulfides are removed
- partial reduction asks which disulfides open first, which remain, and what that pattern reveals about the original topology
That second question is often the more informative one in multi-bridge proteins.
Partial reduction only works when it is deliberate
Random under-reduction is not informative. Controlled partial reduction is.
A useful staged workflow usually defines several variables very tightly:
- reductant concentration
- exposure time
- temperature
- denaturant load
- immediate alkylation or quench after each reduction stage
Without that control, the analyst generates a cloud of intermediates that may be chemically real but analytically unhelpful. With that control, the result becomes a topology ladder. Each rung gives the reader a little more information about which bridges are central, which are exposed, and how the disulfide architecture is organized.
This is also where fragmentation and informatics become essential partners to chemistry. Partial reduction does not replace LC-MS/MS interpretation. It sharpens it. A staged workflow is most powerful when combined with targeted sequencing logic and careful data analysis, because the chemistry is being used to simplify the fragmentation problem rather than to solve it on its own.
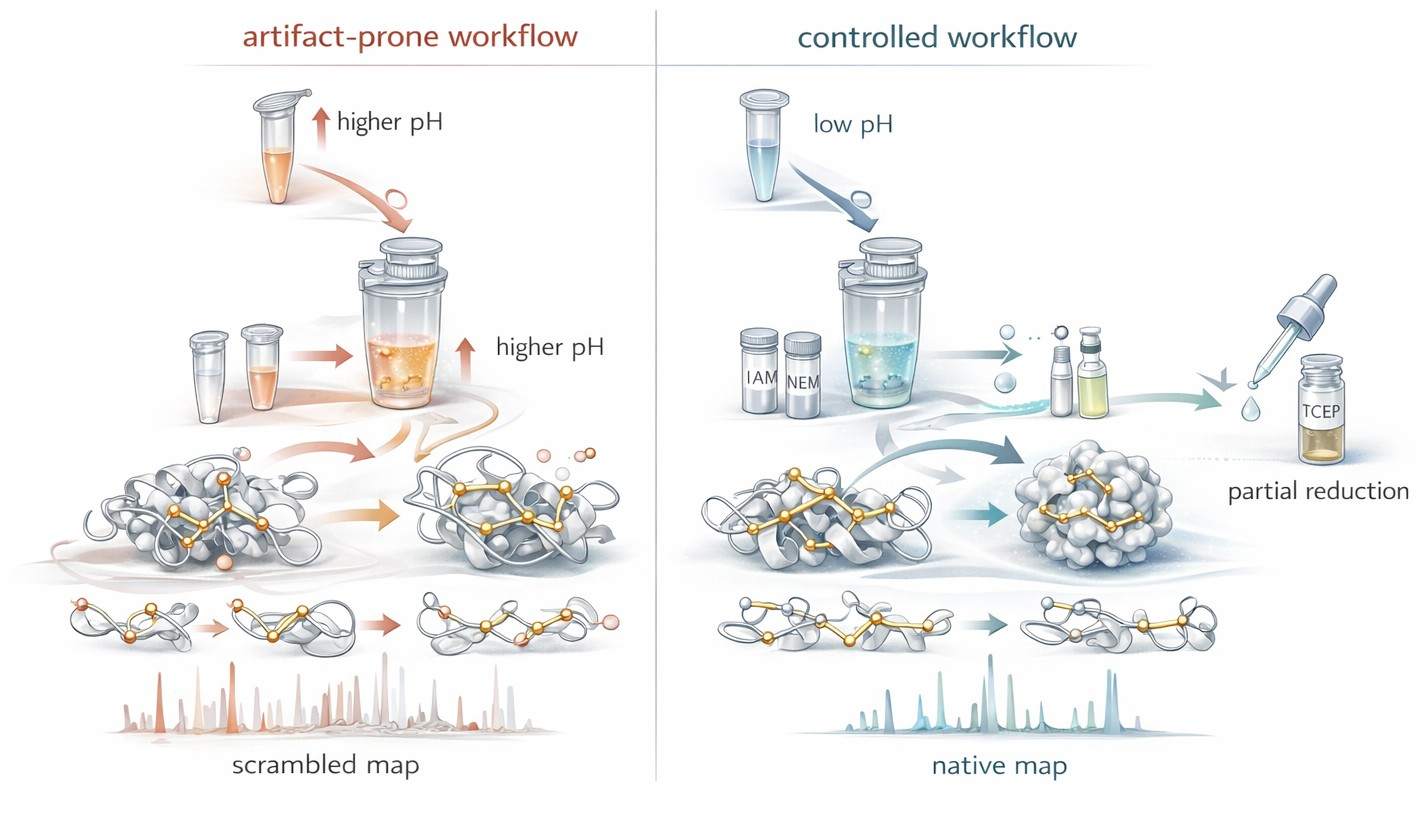 Figure 2: Workflow design to suppress disulfide scrambling
Figure 2: Workflow design to suppress disulfide scrambling
Figure 2 should appear here because the article has now moved from mechanism to active control strategy. The reader has already seen why scrambling happens, why pH matters, why alkylation is a race, and why staged reduction can reveal architecture. The figure should turn those ideas into a clear visual contrast. One side shows the artifact-prone path: permissive pH, delayed trapping, longer exposure, and exchange-driven rewiring. The other shows the topology-preserving path: acidic hold, rapid thiol capture, staged reduction, and immediate control of reactive intermediates. The next section will build on that controlled sample and ask how LC-MS/MS can convert preserved topology into actual evidence.
Bottom-up disulfide mapping starts with linked species
In ordinary peptide-centric proteomics, the basic analytical unit is an isolated peptide. In bottom-up disulfide mapping, the core evidence is different. The most important analyte is often a disulfide-linked species that retains covalent connectivity between two peptide regions after non-reducing digestion.
That difference changes the whole logic of interpretation.
A linked disulfide species carries more than one layer of information at once. It can report which regions of sequence were connected, whether the linkage is intra- or inter-chain, and whether the observed assembly is compatible with the expected native fold. But that value comes with a price. Linked species do not behave like ordinary linear peptides. Their masses reflect combined peptide content. Their fragmentation behavior is more constrained. Their charge-state distributions can shift. Their spectra can quickly become difficult to parse when several cysteines or several linkages are involved.
This is why bottom-up disulfide analysis is not just routine proteomics with one extra search parameter. It is a separate structural problem.
Why disulfide-linked species are analytically special
A disulfide-linked analyte is not just "two peptides stuck together." It is a constrained assembly. That assembly can preserve valuable structural information, but it also changes how the molecule behaves in the mass spectrometer.
At the precursor level, the observed mass reflects the sum of the linked peptide components, adjusted for disulfide formation. In simple cases, that is manageable. In multi-bridge systems, the same logic produces assemblies that are larger, more highly charged, and less predictable than routine peptides. Once several cysteines are present in the same region, the number of plausible pairings rises quickly.
That is why digestion strategy matters so much. The analyst is not only trying to generate peptides. The analyst is trying to generate the right linked intermediates. A digest that looks acceptable from a sequence-coverage perspective may still fail as a disulfide workflow if it does not preserve interpretable linked species.
This is also why bottom-up disulfide analysis often sits close to protein post-translational modification analysis. In both cases, the key challenge is not peptide production alone. The challenge is preserving a chemically meaningful state while turning it into measurable ions.
Fragmentation is where topology becomes evidence
At the precursor stage, the analyst can say that a disulfide-associated species exists. At the fragmentation stage, the analyst begins to defend which linkage is real.
This is the point where many workflows become too generic. They say "perform MS/MS and confirm the bond," as though all tandem spectra answer the same question equally well. They do not. Different fragmentation modes extract different kinds of structural evidence, and that difference becomes critical in disulfide-rich systems.
For small or relatively simple linked species, collision-based fragmentation may be enough to support assignment. But as structural constraint increases, the limitations become clear. The disulfide itself changes the energy landscape of the analyte. Certain backbone cleavages are favored. Others are suppressed. The result can be a spectrum that contains useful ions but still leaves the linkage question partly unresolved.
That is why fragmentation method should be chosen as a structural decision, not a default instrument setting. In disulfide analysis, the best activation method is the one that most directly answers the topology question that remains open after precursor selection.
ETD is valuable because it respects structural constraint
Electron transfer dissociation is especially useful in disulfide work because it often handles constrained linked species more intelligently than standard collision-only logic. In routine peptide analysis, fragmentation is mainly discussed in terms of sequence coverage. In disulfide mapping, the goal is broader. The analyst wants sequence evidence, but also wants linkage-aware evidence.
That is where ETD becomes attractive. It can produce fragmentation behavior that is more compatible with the structural reality of a disulfide-linked analyte. Instead of treating the precursor as a generic peptide ion, it often helps preserve the information needed to see how constrained sequence regions relate to each other.
This is especially helpful when the linked species is the real target of the experiment rather than an inconvenient byproduct. If the goal is to determine which cysteine-containing regions remain connected in a native-like assembly, ETD often provides cleaner evidence than a purely collisional approach.
That does not mean ETD is always the answer. Precursor charge state still matters. Signal quality still matters. Instrument tuning still matters. But in workflows where local linkage assignment is the central problem, ETD is often one of the most useful tools available.
What ETD is especially good at in bottom-up workflows
The practical advantages of ETD in disulfide mapping usually fall into four categories.
Linkage-aware fragmentation
ETD can make it easier to understand how linked peptide regions relate to each other. That does not eliminate interpretation work, but it often improves the analyst's ability to distinguish credible assignments from spectra that only look plausible.
Better preservation of structural context
Many disulfide-rich proteins also carry other modifications. Those may include glycosylation, controlled oxidation states, clipping, or other PTM-level features. ETD is useful when the analyst wants to keep more of that context intact while still collecting informative fragmentation evidence.
More interpretable data from constrained analytes
A linked disulfide species is structurally restricted. ETD is often more compatible with that constraint than a one-size-fits-all collision method. In practical terms, that can mean spectra that are easier to interpret in a topology-aware way.
Strong complementarity with other modes
ETD does not need to replace collision-based MS/MS. In many advanced workflows, the strongest assignments come from combining them. One mode improves local linkage interpretation. The other adds orthogonal sequence support. In difficult samples, that layered evidence is often much more convincing than either mode alone.
UVPD matters when the topology problem becomes whole-protein in scale
Ultraviolet photodissociation is sometimes described too simply as a higher-energy fragmentation option that produces more ions. That description misses the real analytical value. In disulfide-rich proteins, UVPD is powerful because it can generate broad structural evidence from highly constrained analytes, including intact proteoforms that exceed the comfortable range of ordinary bottom-up logic.
The key point is scale. Bottom-up asks which linked peptide regions can be identified after digestion. UVPD-enabled top-down analysis asks what can be learned from the intact protein before the full architecture is broken apart.
That shift matters for several reasons. Some proteins contain multiple bridges that are hard to disentangle after digestion. Some contain several proteoforms whose intact-level context matters as much as the peptide-level assignments. Some contain nested or higher-order disulfide patterns where peptide evidence alone no longer feels complete. In those cases, UVPD becomes valuable not because it is "stronger," but because it is better matched to the size of the topology question.
When bottom-up reaches its limit
Bottom-up disulfide mapping remains the default for good reasons. It is mature, flexible, and often sufficient. But it does have limits, and it is better to state them clearly than to hide them behind general workflow language.
One limit is peptide generation. If non-reduced digestion fails to produce the right linked intermediates, the topology cannot be reconstructed cleanly no matter how strong the instrument is. Another limit is spectral complexity. In multi-bridge proteins, real linked assemblies can become so dense that assignment becomes slow, ambiguous, or overly dependent on software scoring. A third limit is loss of proteoform context. Bottom-up can identify local connectivity while still losing the intact architectural view.
These limits do not make bottom-up weak. They define when escalation becomes rational.
A simple experimental logic helps:
- stay with bottom-up when the digest generates interpretable linked species and the topology is not excessively degenerate
- add ETD when linked-peptide assignments need stronger local structural support
- move toward top-down when intact proteoform context, multi-bridge complexity, or whole-protein topology becomes the central question
That is why the most robust workflows do not treat bottom-up and top-down as competing brands of analysis. They treat them as different structural scales of the same problem.
Top-down disulfide mapping preserves the proteoform question
Top-down analysis begins from a different premise. The intact protein remains the analytical unit. That preserves information that bottom-up necessarily redistributes across peptides.
This changes the questions the analyst can ask. Instead of only asking which peptide regions were connected, the analyst can ask:
- does this intact molecule exist in one dominant disulfide topology or several related states
- do oxidation, clipping, or glycoform differences coexist with specific disulfide architectures
- is the connectivity being observed truly part of the intact proteoform, or only inferred after digestion
That is why top-down disulfide analysis fits naturally with characterization of protein structure and top down-based sequencing. The intact molecule is still the object being read, rather than a puzzle reassembled from peptide fragments.
This is especially useful in analytical characterization workflows for biotherapeutic research, comparability assessment, and structural integrity studies. A molecule may look acceptable at the intact-mass level and still require intact topology-resolved analysis to determine whether the disulfide architecture is truly native.
ETD versus UVPD in practical decision-making
The most useful comparison is not "Which one is better?" The useful comparison is "Which one solves the current ambiguity more directly?"
ETD is often the better first choice when:
- the workflow is bottom-up or middle-down
- linked peptides are visible but not confidently assigned
- the main uncertainty is local connectivity within a constrained analyte
- preserving surrounding modification context is important
UVPD becomes especially valuable when:
- the workflow is intact-protein or top-down focused
- the protein contains multiple disulfides or higher-order topology
- a single precursor needs to yield broad structural evidence
- digestion is likely to simplify away information that matters at the proteoform level
In many projects, ETD and UVPD are not interchangeable. They operate at different structural scales. ETD often sharpens peptide-level linkage evidence. UVPD becomes more attractive when whole-protein topology is the real target.
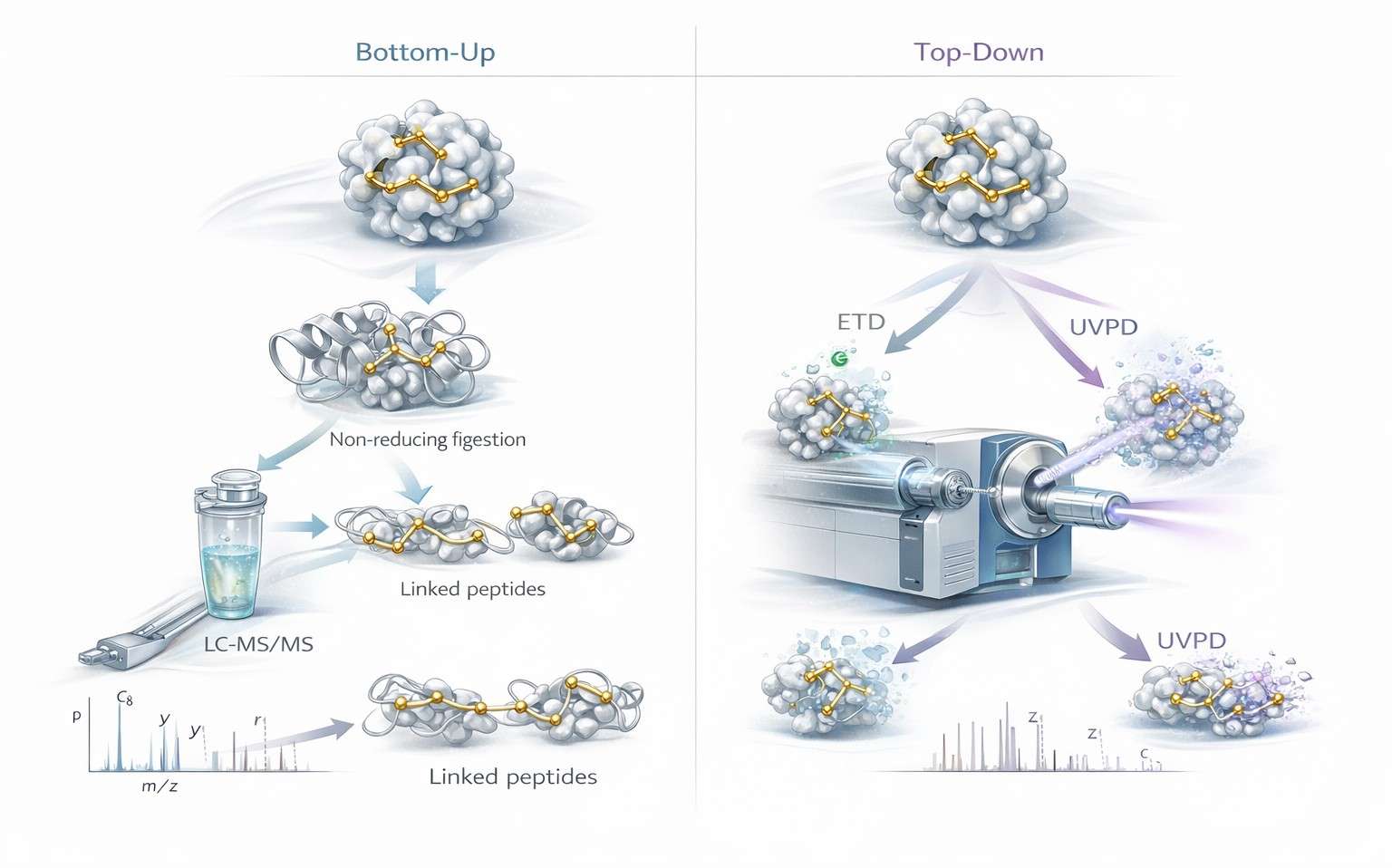 Figure 3: Bottom-up versus top-down disulfide mapping by LC-MS/MS
Figure 3: Bottom-up versus top-down disulfide mapping by LC-MS/MS
Figure 3 should appear here because this is the point where method choice becomes the dominant issue. The figure should let the reader see the difference at a glance. On one side, non-reduced digestion generates linked peptide species that are resolved through bottom-up LC-MS/MS, often with ETD support for local connectivity. On the other side, intact proteins enter top-down workflows where ETD and UVPD interrogate topology at the proteoform scale. The purpose of the image is not just comparison. It is decision support.
Choosing the right workflow
The best disulfide workflow is determined by the protein and by the question being asked.
If the objective is routine confirmation of expected linkages in a tractable protein, bottom-up non-reduced mapping may be enough. If the objective is to troubleshoot low-level heterogeneity, investigate suspected scrambling, or validate ambiguous linked species, bottom-up plus ETD is often the more defensible design. If the objective is to understand proteoform-level structural integrity in a disulfide-rich or heterogeneous molecule, top-down becomes much more attractive.
A practical decision framework looks like this.
Use bottom-up when:
- the digest generates interpretable linked species
- the number of disulfides is moderate
- peptide-scale evidence is enough for the development stage
- sample amount favors peptide-centric analysis
Add partial reduction when:
- several bridges make the linked assemblies difficult to interpret
- staged opening can simplify the topology
- the analyst needs to see which disulfides break first and which remain
Add ETD when:
- collision-based spectra are not decisive
- linked species are constrained and require clearer local evidence
- disulfide assignment must be supported without losing modification context
Move to top-down when:
- intact proteoform context matters
- multiple topological states may coexist
- digestion is removing important structural information
- the problem is whole-protein architecture rather than local linkage alone
The most reliable programs often combine these layers rather than treating them as mutually exclusive. A team may begin with disulfide bond localization, use bottom-up mapping to define likely linkages, then escalate only the ambiguous regions into an intact or top-down workflow. That kind of escalation strategy is often more informative than forcing one platform to answer every question.
DTT, β-ME, and TCEP should be chosen as workflow tools, not as habits
Reducing agents are often discussed as if they were interchangeable. In topology-focused disulfide analysis, they are not. Each one changes the behavior of the workflow differently, especially when the goal is partial reduction, scrambling control, or downstream MS compatibility.
DTT
DTT is familiar and effective for full reduction. It remains common in conventional workflows where complete bond opening is the goal. But it is not automatically the best option for experiments that require carefully staged topology logic. It is most comfortable in standard reduction settings rather than in highly controlled partial reduction design.
β-Mercaptoethanol
β-ME is historically important, but it is usually less attractive in modern precision LC-MS workflows. It can certainly reduce disulfides, yet its handling profile and general workflow convenience often make it a weaker choice for advanced topology-focused work.
TCEP
TCEP is often the most versatile option for advanced disulfide mapping. It is operationally clean, compatible with controlled reduction design, and especially useful when the analyst needs staged opening of multi-bridge architectures. That does not make it universally superior. It makes it especially useful when reduction is being used as an interpretive tool rather than as a simple preparative step.
Experimental design comparison
| Reducing Agent | Best Fit | pH Compatibility | Suitability for Partial Reduction | Quench / Cleanup Considerations | Scrambling-Control Relevance |
|---|---|---|---|---|---|
| DTT | Routine full reduction before conventional peptide workflows | Works well in common reduction conditions but requires controlled downstream handling | Moderate | Needs disciplined follow-up to avoid leaving reactive windows open | Useful, but less ideal when staged topology control is the main goal |
| β-ME | Legacy or simple reduction setups | Serviceable, but less attractive in modern high-control workflows | Low to moderate | Less convenient operationally in MS-centered precision workflows | Usually not preferred when workflow cleanliness is important |
| TCEP | Stepwise reduction and topology-solving workflows | Strong practical fit for controlled reduction design | High | Clean handling and well suited to staged logic with immediate capping | Strong, especially when partial reduction is being used to simplify topology without losing interpretability |
The wrong question is "Which reductant is best?" The better question is "Which reductant creates the cleanest path to the evidence needed in this exact workflow?"
Hidden variables that decide whether a disulfide map is trustworthy
Many disulfide experiments succeed or fail on variables that receive too little attention in short protocols.
Exposure time
Time is not neutral. Every extra minute between denaturation, reduction, alkylation, and quench increases the opportunity for unwanted exchange or reconfiguration.
Local unfolding
A protein does not need to be fully denatured to become topologically vulnerable. Partial opening can expose one sulfur and begin a cascade of new exchange possibilities.
Reagent order
The same reagents can produce different outcomes depending on order of addition. In topology-sensitive workflows, sequence matters as much as composition.
Search strategy
Search space design changes what the software is allowed to believe. A narrow search may miss real linked species. A very broad search may reward attractive but weak assignments. This is where bioinformatics for proteomics becomes especially important, because linked-peptide interpretation is only as strong as the search logic behind it.
Orthogonal confirmation
The strongest assignments rarely depend on one evidence stream. Confidence rises sharply when precursor mass, fragmentation behavior, staged reduction, and intact-level evidence all point to the same topology.
The practical lesson is simple. A trustworthy map is not just acquired. It is engineered.
From bond detection to structural integrity in complex proteomics
This is the real shift in modern disulfide analysis. The highest-value workflows no longer treat disulfide mapping as a narrow exercise in bond localization. They treat it as a structural integrity problem in complex proteomics.
That broader framing changes everything. Reduction and alkylation stop being routine housekeeping and become topology-control steps. Bottom-up and top-down stop competing for attention and become complementary structural scales. Fragmentation choice stops being a default instrument preference and becomes a decision about what kind of structural evidence is still missing.
This is why disulfide analysis increasingly intersects with redox proteomics, protein purity and homogeneity characterization, and advanced structural workflows. A protein can look acceptable in one assay and still carry a topological problem. It can appear oxidized yet not be natively connected. It can appear homogeneous until a topology-preserving workflow reveals hidden rearrangement.
The best modern disulfide strategies are built around one principle: preserve structural truth long enough for mass spectrometry to read it.
Conclusion
Disulfide mapping looks simple when it is reduced to a checklist. Preserve the bonds. Digest the protein. Find the linked peptides. Confirm by MS/MS. In practice, every one of those steps can distort the answer if the workflow is not designed around topology preservation.
The chemistry explains why. Cysteine is reactive. Thiolates drive exchange. pH controls reactivity. Digestion changes exposure. Reduction can reveal structure or erase it. Alkylation only helps if it closes the free-thiol window fast enough. Fragmentation only becomes decisive when it matches the scale of the topology problem.
That is why the strongest workflows are deliberate from start to finish. They use low-pH handling to suppress scrambling. They treat IAM and NEM as kinetic tools, not automatic defaults. They use staged reduction with TCEP when complex architectures need to be opened in sequence. They use ETD when peptide-scale linkage evidence needs stronger support. They move toward UVPD-enabled top-down analysis when intact proteoform context becomes essential.
In the end, disulfide mapping is not just about finding sulfur-sulfur bonds. It is about preserving native topology long enough for LC-MS/MS to measure it honestly.
FAQ
What is the main cause of artificial disulfide scrambling during LC-MS sample preparation?
The main cause is free-thiol-mediated exchange. Once cysteine residues become reactive thiolates under permissive conditions, they can attack native disulfides and generate non-native linkages.
Why is low pH used in disulfide-preserving workflows?
Low pH suppresses thiolate formation. That reduces thiol-disulfide exchange and helps preserve the native disulfide state during handling and digestion.
Is IAM always better than NEM for thiol capping?
No. IAM is common and convenient for downstream database searching, but NEM may be a better choice when rapid thiol trapping is the main priority and scrambling suppression is critical.
Why use partial reduction instead of full reduction?
Partial reduction can reveal complex disulfide topology in stages. It helps the analyst see which bridges open first and how the native architecture was organized.
When does non-reduced digestion become unreliable for multi-bridge proteins?
It becomes less reliable when digestion produces overly complex linked assemblies, when exposure windows allow exchange, or when the resulting spectra no longer support confident topology assignment without staged simplification.
When should ETD be preferred over collision-based fragmentation?
ETD is especially useful when the analyte is a constrained disulfide-linked species and the main question is local connectivity rather than routine sequence coverage.
When should UVPD or top-down analysis be considered?
UVPD-enabled top-down analysis becomes attractive when intact proteoform context matters, when several disulfide topologies may coexist, or when digestion removes too much structural information.
Can LC-MS alone prove native disulfide connectivity?
Not always. LC-MS can strongly support native connectivity, but the highest-confidence assignments usually come from combining precursor mass, fragmentation, controlled sample preparation, and, when needed, intact-level or orthogonal confirmation.
Which reducing agent is best for advanced disulfide mapping?
There is no universal best choice. DTT works well for routine full reduction, β-ME is generally less convenient in modern precision workflows, and TCEP is often the most useful option for controlled or partial reduction strategies.
References
- Wang Y, Xu G, Guo Z. Top-down mass spectrometry for disulfide bond characterization in biotherapeutic proteins. TrAC Trends in Analytical Chemistry. 2023;164:117104. DOI: 10.1016/j.trac.2023.117104
- Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG, Loo JA. An integrated native mass spectrometry and top-down proteomics strategy for higher-order disulfide mapping. Analytical Chemistry. 2024;96(5):2210-2219. DOI: 10.1021/acs.analchem.3c04172
- Zhang X, Chen W, Ge Y. Top-down mass spectrometry for structural characterization of intact proteins with disulfide bonds. Mass Spectrometry Reviews. 2024;43(2):e21888. DOI: 10.1002/mas.21888
- Srebalus Barnes CA, Lim MY, Godinho JM, et al. Top-down analysis of disulfide-linked proteins using photoactivation and electron-based dissociation strategies. Journal of the American Society for Mass Spectrometry. 2020;31(9):1839-1849. DOI: 10.1021/jasms.0c00179
- Robinson AS, Bulleid NJ. Protein disulfide isomerase and oxidative protein folding in the endoplasmic reticulum. Nature Reviews Molecular Cell Biology. 2020;21(1):20-37. DOI: 10.1038/s41580-019-0153-3
- Bulleid NJ. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold Spring Harbor Perspectives in Biology. 2012;4(11):a013219. https://cshperspectives.cshlp.org/content/4/11/a013219.full.pdf
- Gardner MW, Freitas MA. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. Journal of Proteome Research. 2008;7(1):138-144. https://europepmc.org/articles/PMC2749473
- Yang Y, Franc V, Heck AJR. Disulfide bond analysis in proteins and peptides by mass spectrometry. Mass Spectrometry Reviews. 2017;36(4):532-556. DOI: 10.1002/mas.21509
- Oka OBV, Bulleid NJ. Forming disulfides in the endoplasmic reticulum. Biochimica et Biophysica Acta - Molecular Cell Research. 2013;1833(11):2425-2429. DOI: 10.1016/j.bbamcr.2013.02.007









