COVID-19 caused by SARS-CoV-2 from the end of 2019 is highly infectious. The disease has progressed to a global pandemic. Understanding the pathogenesis of SARS-CoV-2 and developing drugs to treat SARS-CoV-2 infection is a priority in research.
Here are three case studies.
Case 1 Proteomics fuels research on COVID-19 pathogenesis (1)
Early detection and effective treatment of patients with severe COVID-19 remains a major challenge. In this study, TMTpro (16plex) labeling technology combined with liquid-quality concatenated assays and non-targeted metabolomics were used to sample and analyze patients with severe COVID-19, patients with mild COVID-19, patients with suspected COVID-19, and healthy populations. 894 proteins and 941 metabolites were identified and quantified.
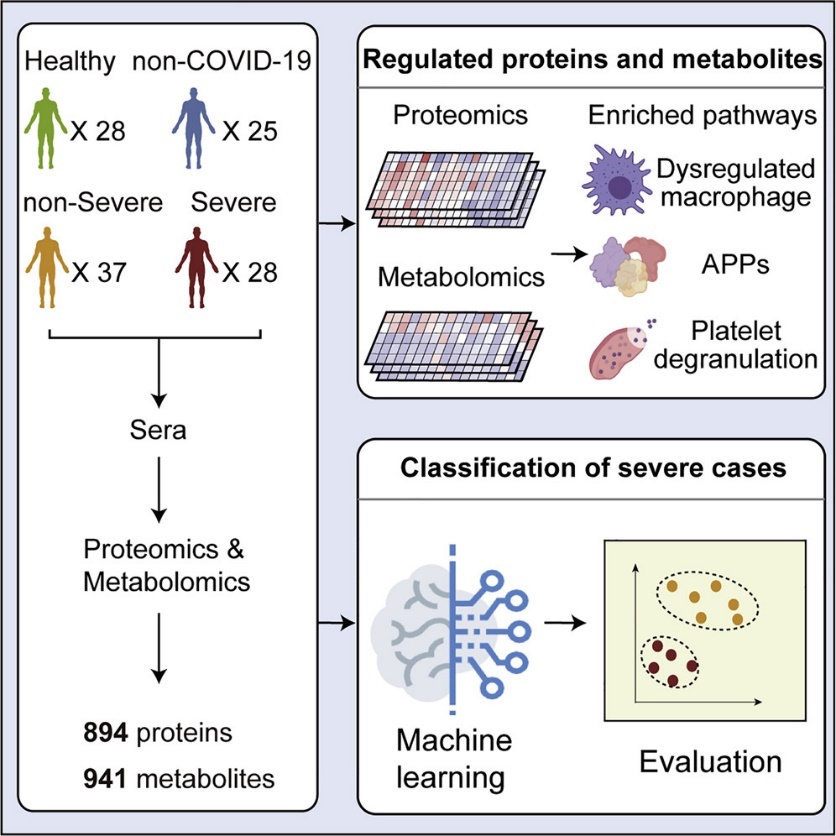
Proteomic and Metabolomic Characterization of COVID-19 Patient Sera (Shen et al., 2020)
The potential of proteins and metabolites in identifying which COVID-19 patients are likely to develop severe disease is illustrated by the analysis of a panel of serum proteins and metabolites. These data provide a panoramic view of the molecular changes in the blood caused by SARS-CoV-2 infection and provide useful diagnostic and therapeutic clues to combat the spread of COVID-19.
Case 2 DIA-based proteomic analysis of nasopharyngeal swabs from COVID-19 patients (2)
A major drawback of the DDA method is the lack of reproducible measurements across samples, mainly due to the random nature of precursor ion sampling during MS/MS analysis. In contrast, the DIA method ensures that all precursor ions are fragmented within any predetermined isolation window to significantly improve the reproducibility of proteomics measurements.
The authors of this study designed a mass spectrometry study supporting the PASEF mode coupled with DIA. The performance of DDA and DIA run in PASEF mode was assessed by injecting peptide digests from cultured Jurkat cells, and then the PASEF-DDA data obtained were analyzed. From the three DDA replicate experiments, 4747 proteins were identified and 2721 proteins (57%) were detected in replicates. In contrast, the same 4490 (94%) proteins were identified in each of the three DIA experiments.
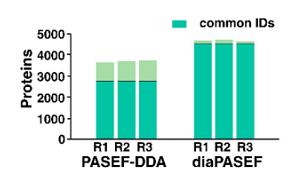
Bar charts of the number of protein groups from triplicates of PASEF-DDA and diaPASEF experiments (Mun et al., 2021)
Proteome unbiased measurements were performed on single SARS-CoV-2 positive and negative NP swab samples by injecting the same amount of peptides (i.e. 1 μg) in diaPASEF mode using an isolation window of 25 m/z. A total of 79,703 peptides corresponding to 5,023 proteomes were detected, with an average of 3,387 proteomes detected per sample.
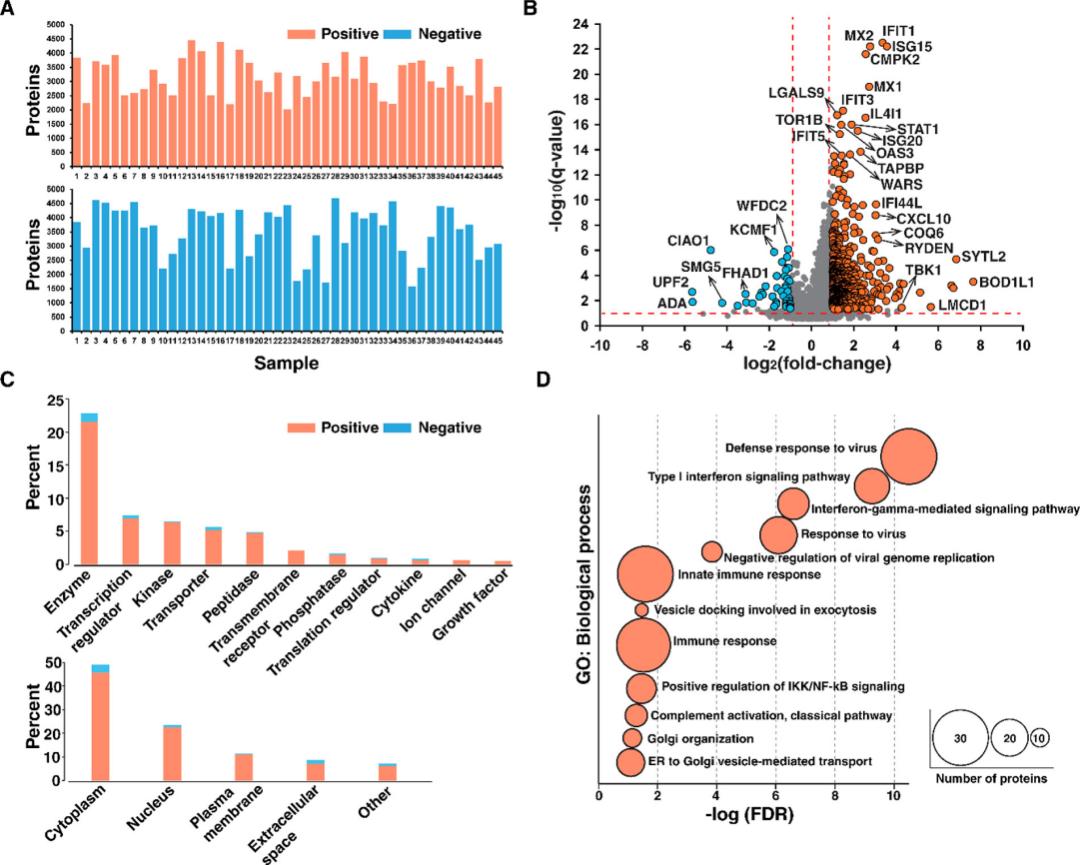
Quantitative analysis using diaPASEF (Mun et al., 2021)
This study demonstrates that comprehensive proteomic analysis of clinical samples using the diaPASEF approach can help decipher the host response to SARS-CoV-2 infection. The data presented herein reveal the activation of several biological processes in SARS-CoV-2 positive subjects, including the innate immune response (IFN signaling pathway), as well as viral replication (cytokinesis and ER/Golgi transport).
Case 3 Proteomics involvement in therapeutic selection for SARS-CoV-2 infection(3)
To identify pathways associated with viral pathogenicity and potential drug targets, this study performed molecular assays on infected cells by an unbiased proteomics approach. The study used SARS-CoV-2 single infected Caco-2 cells cultured for 2-24 hours and analyzed for protein differences by coupling two labeling methods, stable isotope labeling (SILAC) and tandem mass labeling (TMT).
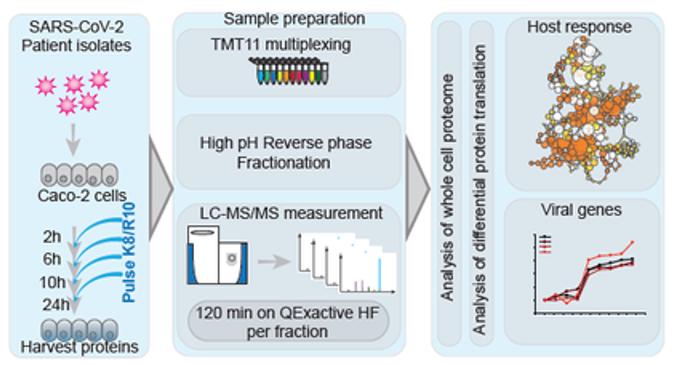
Experimental scheme for translatome and proteome measurements (Bojkova et al., 2020)
The SARS CoV-2 virus was first incubated with Caco-2 cells for a certain period of time. Two hours prior to sample collection, the medium was changed to relabeled SILAC medium to label the newly synthesized proteins. The proteins with partial SILAC labeling were enzymatically cleaved into peptides, labeled by TMT11plex reagent and graded by high-pH reverse phase. Finally, high-throughput proteomics analysis was performed with the aid of Easy nLC 1200-Q Exactive HF high-resolution mass spectrometry platform to characterize the host and viral gene expression changes.
In this study, quantitative proteomic analysis of SARS-CoV-2 infection on host cells identified the host cellular pathways regulated by SARS-CoV-19 infection and revealed relevant target pathways for drug inhibition of virus replication in human cells.
You may be interested in:




Resources
- Shen, B., Yi, X., et al. (2020). Proteomic and metabolomic characterization of COVID-19 patient sera. Cell, 182(1), 59-72.
- Mun, D. G., Vanderboom, P. M., et al. (2021). DIA-based proteome profiling of nasopharyngeal swabs from COVID-19 patients. Journal of Proteome Research, 20(8), 4165-4175.
- Bojkova, D., Klann, K., et al. (2020). SARS-CoV-2 infected host cell proteomics reveal potential therapy targets.




