More mass spectrometry based strategies are emerging and applied to proteomic research as the technology continues to be developed. Methods of proteomics using mass spectrometry can qualitatively analyze and identify proteins. For example, protein identification by peptide mass fingerprinting is quick, sensitive, and high-throughput; peptide sequence tagging can identify proteins in mixtures. With the development of proteomics and the corresponding technologies, measurement on the change in protein abundance has attracted more attention. Mass spectrometry based label-free quantitative method is important for detecting protein abundance. This type of method is characterized by high throughput, high selectivity, high sensitivity, wide dynamic range, and good repeatability. At present, the commonly used label-free quantitative methods include selective / multiple reaction monitoring and parallel reaction monitoring.
Selective / Multiple Reaction Monitoring Mass Spectrometry
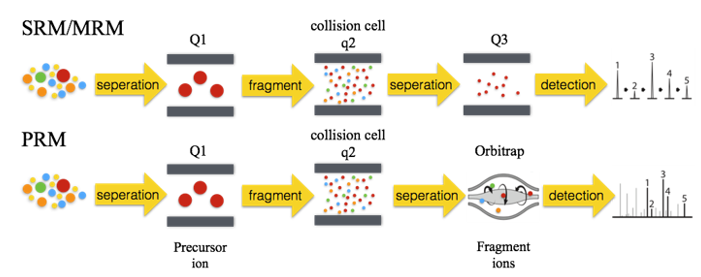
Selective reaction monitoring (SRM) is a technique of targeted sample quantification based on measured values or information assumed according to the principles in mass spectrometry detection. The main instrument is the triple quadrupole mass spectrometer. The first and third quadrupole rods are used as mass filters to specifically select peptide ions (precursor ions) and fragment ions (product ions) with the same pre-set mass charge ratio. The second quadrupole is used as a collision cell, usually using collision-induced dissociation (CID) to fragment the precursor ions. The two mass selections make the SRM technique highly selective. In addition, background noise is reduced, interfering ions removed, sensitivity improved, and the determination of low abundance proteins in complex samples became feasible. On the premise of ensuring data quality, multiple reaction monitoring (MRM) was developed to maximize the number of tests performed during each run. MRM allows the simultaneous monitoring of multiple selective reactions.
Parallel Reaction Monitoring Mass Spectrometry
Parallel reaction monitoring (PRM) is derived from MRM, which can also simultaneously perform relative or absolute quantitative detection of multiple target proteins in complex biological samples. PRM is based on high-resolution, high-precision mass spectrometers (such as Q-Exactive HF-X, etc.). It uses selective detection capability of a quadrupole mass analyzer to selectively detect the precursor ion information of the target peptide. The precursor ions are then fragmented in the collision cell. Finally, a high-resolution, high-quality precision Orbitrap analyzer is used to detect all fragments in the selected precursor ion window in the secondary mass spectrum.
Comparing Selective / Multiple Reaction Monitoring and Parallel Reaction Monitoring
- Similarities
The principle of screening target proteins in the first-stage mass spectrometry of these technologies is the same, and the precursor ions are screened through the quadrupole for the next step of detection.
- Differences
1. MRM detects specific product ions after the fragmentation of the precursor ions, and qualitatively quantifies the protein through the one-to-one correspondence between the precursor ion and the product ion. PRM detects all the product ions through a high-resolution detector after the precursor ions are fragmented.
2. Due to the different detection modes of secondary mass spectrometry, PRM is more accurate than MRM, and with much less ion interference.
3. PRM is more convenient. Compared with MRM, the mass-to-charge ratio and charge number of the precursor ion are the only parameters needed when using PRM. Finding an accurate precursor ion-daughter ion pair for detection is unnecessary.
References
- Prakash A, Rezai T, et al. Interlaboratory reproducibility of selective reaction monitoring assays using multiple upfront analyte enrichment strategies. Journal of proteome research, 2012, 11(8): 3986-3995.
- Bourmaud A, Gallien S. Parallel reaction monitoring using quadrupole‐Orbitrap mass spectrometer: principle and applications. Proteomics, 2016, 16(15-16): 2146-2159.






