Proteome is the sequence identification and content analysis of all proteins in a living organism, but the information of PTM (post-translational modification) cannot be ignored. Phosphorylation proteins account for about 1/3 of all proteins in organisms, so phosphorylation has become one of the types of protein PTMs that have received the most attention.
Common phosphorylated protein identification methods
Principle: The protein separated by SDS-PAGE gel electrophoresis is transferred from the PAGE gel to the solid phase carrier, and the primary antibody is used as a probe, combined with a labeled secondary antibody, and then developed with a chromogenic reagent. According to the specificity of antigen-antibody binding, only the phosphorylated protein will appear as a band at the corresponding molecular mass.
Features: Strong specificity and high resolution, but only semi-quantitative, and now mostly used to verify mass spectrometry results.
Disadvantages: It is difficult to identify new sites and difficult to detect multiple phosphorylation sites of proteins,due to the difficulty of preparing specific phosphorylated antibodies.
Principle: Using radioisotope 32P-labeled phosphate as the phosphate group donor, the phosphate group with 32P labeling is transferred to the substrate protein under kinase catalysis and then separated by gel electrophoresis, and the phosphorylated protein is detected by radioactive autoradiography.
Features: It can detect protein phosphorylation sites and single protein phosphorylation.
Disadvantages: Highly radioactive and dangerous.
Principle: The mass spectrometer can ionize the analyzed sample into charged ions. These ions are separated in space or time under the action of an electric or magnetic field. After detection, the mass-to-charge ratio (m/z) and relative intensity of the mass spectrum are obtained. To calculate the mass of the molecules in the analyte, the mass spectrogram can be used for qualitative analysis of the analyzed substances, and quantitative analysis can be done according to the ionic strength. Since the molecular weight of the phosphorylated polypeptide will increase by 79.9663 Da, the sequence-level phosphorylation modification site can be inferred from other fragment ion signals.
Features: High throughput, thousands of proteins can be obtained simultaneously for qualitative and quantitative information, and multiple phosphorylation modification sites and levels of the same protein can be determined.
Disadvantages: High technical difficulty, relying on professional platform and rich knowledge of mass spectrometry.
Phosphorylated Protein Identification Method
Phosphorylated protein "bottom-up" mass spectrometry workflow:
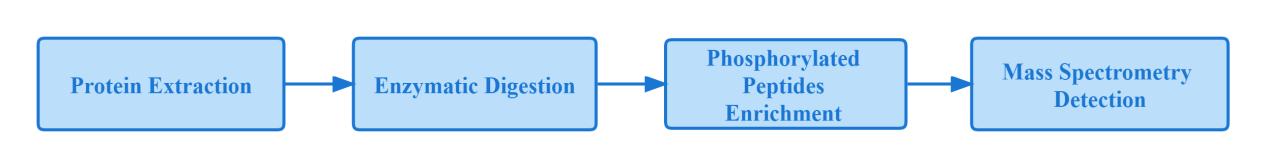
Since protein phosphorylation is a dynamic process, it is not easy to detect low abundance of phosphorylated proteins at low levels in cells, and high levels of phosphorylated peptides not only inhibit phosphorylated peptide ionization, but also obscure phosphorylated peptide signals. Therefore, effective enrichment of purified phosphorylated proteins becomes critical before performing mass spectrometry analysis.
Phosphorylated protein enrichment method
Considering the enrichment capacity and cost, the most widely used ones include immobilized metal ion affinity chromatography (IMAC) and titanium dioxide affinity chromatography (TiO2).
IMAC principle: IMAC is mainly composed of three parts: metal ion, chelating agent and matrix carrier. Under acidic conditions, the chelating agent on the stationary phase forms a coordination compound with the metal ion, and the metal ion is immobilized on it. The positively charged metal ion combines with the negatively charged phosphate group to adsorb the phosphorylated peptide segment.
Principle of TiO2: TiO2 belongs to a metal oxide with strong enrichment ability in metal oxide affinity chromatography (MOAC), although both and IMAC are based on the electrostatic interaction between metal ions and phosphorylated peptides to purify and enrich phosphorylation protein. However, due to the strong interaction of metal atoms and oxygen atoms and more stable and metal atoms are not easy to lose, TiO2 is widely used.
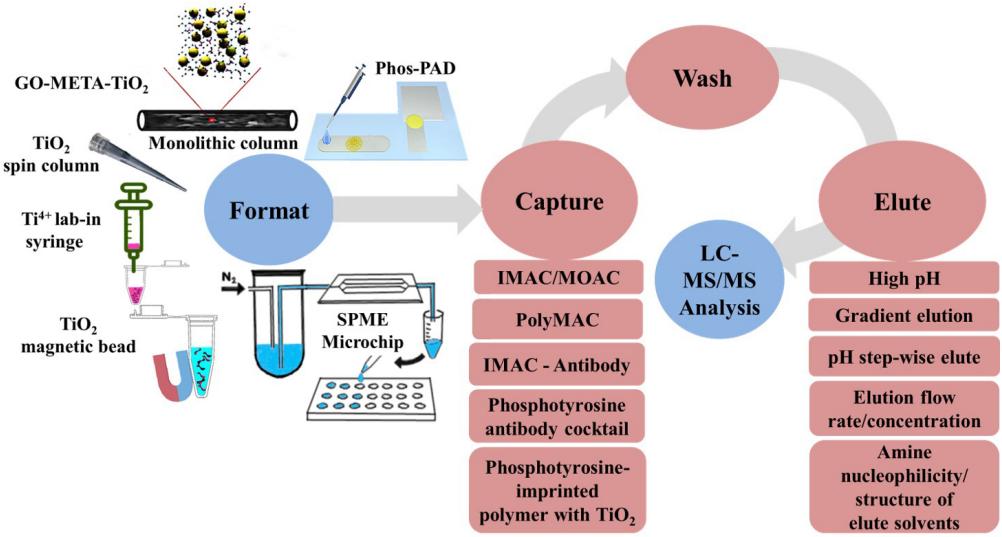
Phosphopeptide enrichment workflow
Phosphorylated Protein Quantitative Methods
The enriched phosphorylated peptides are separated by chromatography and can be detected by mass spectrometry. Commonly used mass spectrometry quantification methods include TMT, LFQ and DIA.
TMT is a quantitative technique that adds isotope labeling, which can simultaneously label and analyze 16 samples with high accuracy. In addition to the identification of the whole proteome, TMT can also be used for the study of PP. However, each labeling reagent can label a maximum of 100ug of peptides. In order to achieve an ideal enrichment efficiency, it is generally recommended that the initial amount of protein phosphorylation modification enrichment should not be less than 1mg and if TMT labeling quantitative method is used, it is recommended that the number of samples should be at least 10. Therefore, the TMT labeled quantification method requires high sample volume and is more expensive, while non-labeled quantification is more suitable for large-scale assays.
LFQ
Compared with TMT-labeled quantification, non-labeled quantification is simpler to operate, less sample loss, and more proteins can be quantified in one experiment, but it is heavily dependent on the stability of the mass spectrometer. The traditional label-free quantification method is LFQ (Label-free), which relies on the data acquisition mode of DDA, while phosphopeptide isomers are difficult to sample and assign with DDA. Systematic and reproducible analysis of phosphorylation sites in a large number of samples is challenging.
DIA
Can DIA acquisition mode be used to analyze phosphopeptides?
Since the DIA data acquisition window is wider and the spectra are more complex, the ability to correctly locate phosphorylation sites in the mixed spectra and to correctly analyze phosphopeptide isomers requires a higher level of spectral processing.
In February 2020, a research team from Denmark broke through this technical difficulty and published an article in Nature Communications, proposing a complete workflow for DIA and direct DIA (DIA without DDA library building) to identify phosphorylated proteins. The research team enriched phosphorylated peptides in wild-type yeast cells by TiO2 and then quantified them by DDA and DIA and dDIA, respectively. The optimized DIA method was found to quantify up to twice as many phosphorylated peptides as DDA, and dDIA identified more than DDA with better reproducibility. Yeast phosphopeptide was mixed with HeLa cell phosphopeptide samples in different proportions, and then quantified separately. The results showed that the peptides quantified by DIA and dDIA also increased proportionally with the increase of concentration and their accuracy was better. DIA and DDA have comparable error rates, but DIA is superior to DDA in locus coverage, sensitivity, and dynamic range.
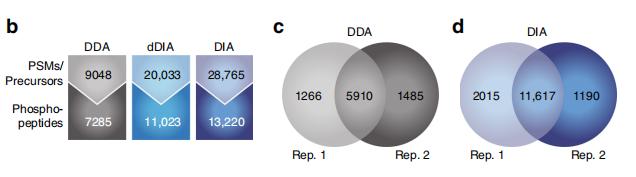
DDA, DIA and dDIA phosphorylated protein assay results
Summary
TMT can be used for phosphorylated protein detection if funds are sufficient and the sample size is small, otherwise the DIA method is recommended, especially in the case of large sample size, DIA is better than LFQ in terms of reproducibility and accuracy.
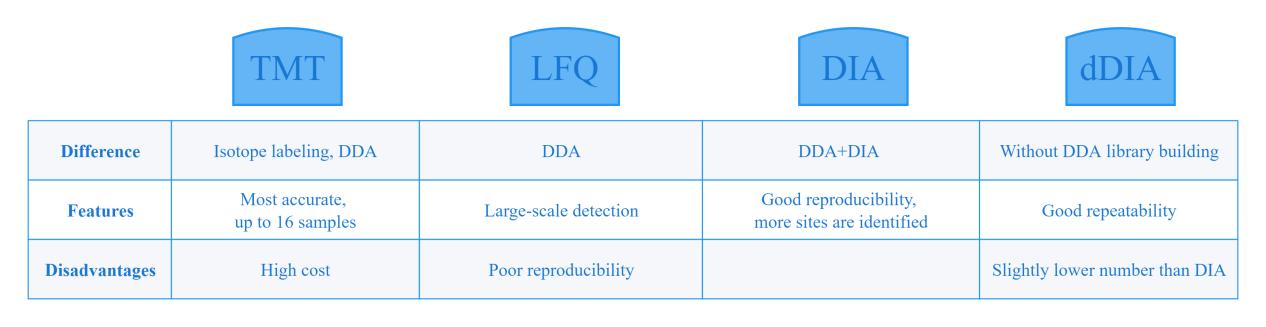
Comparison of phosphorylated protein quantification methods
References
- Needham E J, Parker B L, Burykin T, et al. Illuminating the dark phosphoproteome[J]. Science signaling, 2019, 12(565).
- Harsha H C, Pandey A. Phosphoproteomics in cancer[J]. Molecular oncology, 2010, 4(6): 482-495.
- Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma[J]. Nature, 2019, 567(7747): 257-261.
- Mergner J, Frejno M, List M, et al. Mass-spectrometry-based draft of the Arabidopsis proteome[J]. Nature, 2020, 579(7799): 409-414.
- Bonne Køhler J, Jers C, Senissar M, et al. Importance of protein Ser/Thr/Tyr phosphorylation for bacterial pathogenesis[J]. FEBS letters, 2020.
- Xu N, Luo X, Wu W, et al. A Plant Lectin Receptor-like Kinase Phosphorylates the Bacterial Effector AvrPtoB to Dampen Its Virulence in Arabidopsis[J]. Molecular Plant, 2020, 13(10): 1499-1512.
- Brüning F, Noya S B, Bange T, et al. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation[J]. Science, 2019, 366(6462).
- Bekker-Jensen, D.B., Bernhardt, O.M., Hogrebe, A. et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat Commun 11, 787 (2020).
- W. Qiu, C. Evans, A. Landels, T.K. Pham, P.C. Wright, Phosphopeptide enrichment for Phosphoproteomic Analysis - A Tutorial and Review of Novel Materials, Analytica Chimica Acta.






