The method of peptide and protein de novo sequencing by mass spectrometry
De novo sequencing is a method to analyze and identify peptide sequences and some post-translational modified proteins. Different from some other analysis methods, which depend on a known protein sequence database or a known mass spectrometry database, de novo sequencing uses tandem mass spectrometry for direct analysis based on the fragmentation characteristics of the peptide. Hence, peptide sequences that are not included in the protein database, protein sequence of new species and those whose genome has not been sequenced can all be analyzed. The software developed by this method includes PepNovo, PEAKS, and pNovo.
The basic principle of peptide and protein de novo sequencing by mass spectrometry
In mass spectrometry, the particles of the substance to be analyzed are first ionized before passing through the electromagnetic field. Since ions with different mass-to-charge ratios are subject to different forces in the electromagnetic field, their movement trajectory and flight time, features that can separate and detect ions, are different. In the process of peptide identification using tandem mass spectrometry, peptide de novo sequencing can directly infer the most likely peptide sequence from the secondary map. After finding a specific fragmentation pattern, the corresponding amino acid information can be calculated based on the mass difference between the mass spectrum peaks and the amino acid Post-translational modification.
The workflow of peptide and protein de novo sequencing by mass spectrometry
The workflow of protein/peptide de novo sequencing is as follows.
1. Extracting the protein to be analyzed from the sample tissue using biochemical fractionation or affinity selection process. Use relevant enzymes (enzymolysis), such as trypsin, to generate peptides.
2.Isolate and purify the peptide digestion product using high-performance liquid chromatography.
3. Before entering the ion source, separate and wash the peptides by high pressure liquid chromatography.
4. The peptide passes through the ion source and is converted into highly charged droplets. After dehydration, it enters the mass spectrometer to generate the first-level mass spectrum.
5. A priority list of the peptide is generated by the computer.
6. The peptide is selected for the next round of fragmentation. The mass-to-charge ratio of the ions can be obtained by detecting the ion movement in the electromagnetic field to form a tandem mass spectrometer;
7. During the analysis of the mass spectrometer, peptide ions with a specific mass-to-charge ratio pass through a certain mass;
8. This kind of energy bombardment cleavage can be deduced according to the mass-to-charge ratio information of these fragmented ions, and the corresponding peptide amino acid residues can be deduced from the arrangement and combination, so as to resolve the peptide sequence corresponding to the mass spectrum;
9. Software is used to analyze the secondary mass spectrum peaks of each peptide. Splicing the peptide sequence to obtain full-length protein sequence.
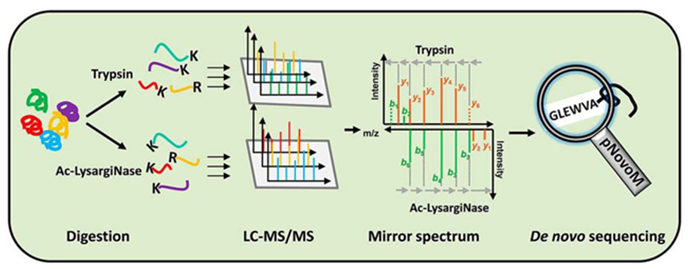
Figure 1. The process of peptide de novo sequencing by mass spectrometry (Hao, et al. 2019).
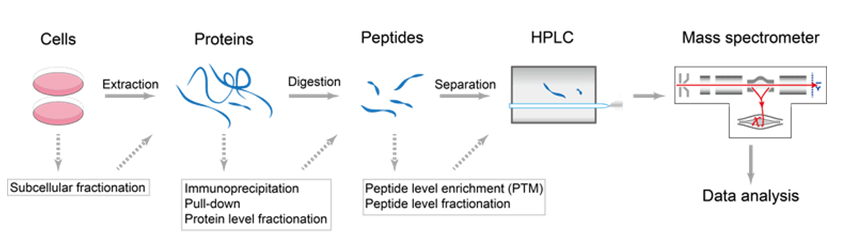
Figure 2. The process of de novo sequencing by the conjunction of HPLC and mass spectrometry
The applications of peptide and protein de novo sequencing by mass spectrometry
Some applications include analysis and detection of unknown peptide sequences in biological samples, detection of peptides with missing terminal residues, identification of lysine and leucine, N-terminally blocked and cyclic proteins identification, accurate determination of the sequence information of unknown proteins or peptides to facilitate the functional study of unknown proteins, accurate determination of the sequences of commercially modified proteins and enzymes, accurate determination of the sequences of proteins expressed in stable cell lines, and monoclonal analysis of antibody primary structure sequences and so on.
References
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature, 2003,422(6928):198-207.
- Hao Yang, Yan Chang Li, Ming Zhi Zhao, et al. Precision De Novo Peptide Sequencing Using Mirror Proteases of Ac-LysargiNase and Trypsin for Large-scale Proteomics. Molecular & cellular proteomics: MCP, 2019.
- Medzihradszky K F, Chalkley R J. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrometry Reviews, 2013, 34(1).
- Marshall Bern, Yuhan Cai, David Goldberg. Lookup Peaks: A Hybrid of de Novo Sequencing and Database Search for Protein Identification by Tandem Mass Spectrometry. Analytical Chemistry, 2007, 79(4):1393-1400.





