What is Label-Free Quantification Technique?
The protein label-free quantification technique analyzes protein enzymatic peptide fragments by liquid-quality coupling and compares the signal intensity of the corresponding peptide fragments in different samples for relative quantification of the protein. This method is not dependent on isotope labeling and does not require expensive stable isotope labels as internal reference. The technique is now widely used in the fields of disease marker screening, disease development mechanism research, and drug action target research.
DDA vs. DIA Label-Free Quantitative Proteomics
- DDA-based MS1 label-free quantitative proteomics
Each sample is first enzymatically cleaved into a peptide. The samples were then subjected to LC-MS/MS analysis by data-dependent acquisition (DDA). Scanning for DDA includes a high resolution MS1 full scan, followed by quadruple rod/ion trap for isolation of parent ions for specific conditions, collision cell fragmentation and MS2 fragmentation ion spectrogram acquisition. The ion flow chromatogram of the extracted high resolution parent ion is used as a quantitative feature and MS2 is used for peptide sequence identification.
The method is based on the primary spectrum parent ion intensities, such as spectral peak height, spectral peak area and spectral peak volume information for protein quantification. In the primary mass spectra, each parent ion is an ionized peptide, including three-dimensional information such as liquid chromatography retention time, mass-to-charge ratio, and ion intensity. The parent ion signal intensity is correlated with the ion concentration. Therefore, the ion peak intensity (peak height, peak area and peak volume, etc.) corresponding to the identified peptide is extracted from the primary spectrum to reflect the abundance of the peptide.
Based on this strategy, MS1 quantification and MS2 identification are independent of each other, allowing peptide identification information to be transferred throughout the sample dataset and facilitating the quantification of low-abundance peptides. The MS1 quantification strategy is often used for large-scale quantitative proteomics studies.
- DIA-based label-free quantitative proteomics
Data independent acquisition (DIA) is performed by MS/MS fragmentation of all peptide parent ions in a specific mass-to-charge ratio (m/z) range indiscriminately after a high-resolution full scan of the primary mass spectrometry. In DIA, high-resolution MS2 spectra are used for peptide identification, and both high-resolution MS1 and MS2 can be used for peptide/protein quantification.
Compared with the MS1-based quantification method, the advantages of the DIA method are (1) non-discriminatory access to all peptides without loss of information of low-abundance proteins, as shown by fewer missing values; (2) fixed cycle time, uniform number of scan points, and high quantification accuracy, as shown by a lower coefficient of variation (CV) in the intensity of quantified proteins; (3) a lower coefficient of CV); (3) no randomness in the selection of peptides, and the data can be retraced; (4) better reproducibility for complex protein samples, especially low-abundance proteins.
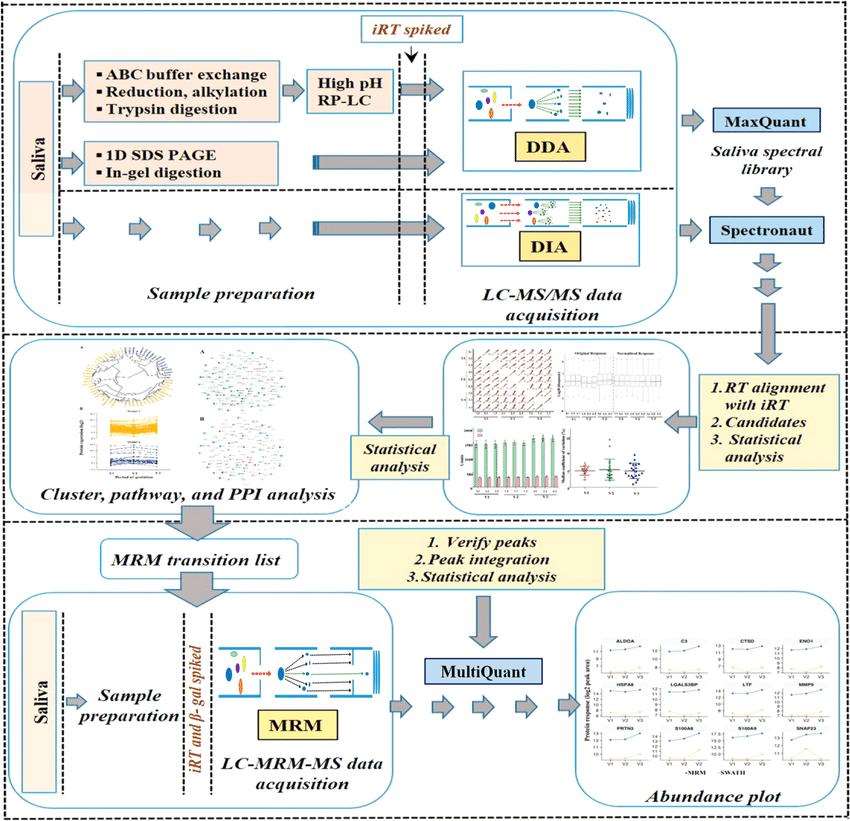
Schematic representation of the label-free quantitative discovery and MRM based targeted validation workflow (Dey et al., 2020).
What is the difference in label based and label free quantitative proteomics?
| Label-based quantification (iTRAQ/TMT) | Labe-free quantification | |
|---|---|---|
| Throughput (number of protein identifications) | High | Low |
| Number of samples detected | Limited number of tags | Less restriction, suitable for large sample size |
| Amount required for the original sample | High | Low |
| Detection of the presence or absence of protein | -- | Detectable |
| Sample parallelism requirement | Good parallelism, high reproducibility of experiments | High requirement for parallelism and low reproducibility of experiments |
| Sample requirements | Similar sample background (same species and same sample type) | Samples from different species, sources or sites require simultaneous proteomic analysis |
| Experimental error | Labeling and liquid phase grouping can introduce experimental error | No error is introduced |
| Modified omics | Only suitable for phosphorylation modification projects | General modification omics is possible |
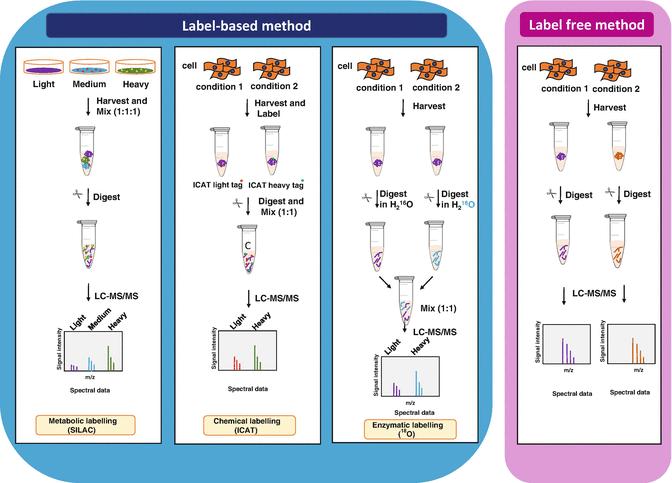
General scheme of quantitative proteomics (Anand et al., 2017)
You may be interested in:

Reference
- Dey, A. K., Kumar, B., et al. (2020). Salivary proteome signatures in the early and middle stages of human pregnancy with term birth outcome. Scientific reports, 10(1), 1-15.
- Anand, S., Samuel, M., Ang, C. S., Keerthikumar, S., & Mathivanan, S. (2017). Label-based and label-free strategies for protein quantitation. In Proteome bioinformatics (pp. 31-43). Humana Press, New York, NY.




