The dynamic changes of proteome abundance in cells have an important impact on various life processes. For example, the occurrence and development of many diseases is often accompanied by abnormal expression of certain proteins. Quantitative proteomics is the accurate quantification and identification of all proteins expressed in a genome or all proteins in a complex mixed system. At present, quantitative proteomics technology is mainly divided into labeling strategies and non-labeling strategies, in which the labeling strategies are divided into in vivo labeling (such as SILAC), and in vitro labeling (such as iTRAQ, TMT).
Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)
iTRAQ is a quantitative proteomics research technology developed by AB SCIEX company in 2004. This technique enables comparison of proteins in many different samples. For example, it can study differences in protein expression levels in tissue samples under different pathological conditions or different developmental stages. At the same time, iTRAQ can accurately quantify and identify all proteins expressed in one genome or all proteins in a complex mixed system. iTRAQ can use 8 different isotope reagents to simultaneously label and compare 8 different protein samples. These reagents are composed of report group, balance group and amine-specific reactive group.
- The workflow of iTRAQ
The general process of iTRAQ technology is shown in the figure 1. Briefly, samples are usually cleaved by trypsin, alkylated, and enzymatically digested into peptides. The generated peptides are differentially labeled with multiple tags of iTRAQ reagent before mixing together labeled samples to be analyzed by LC-MS/MS.
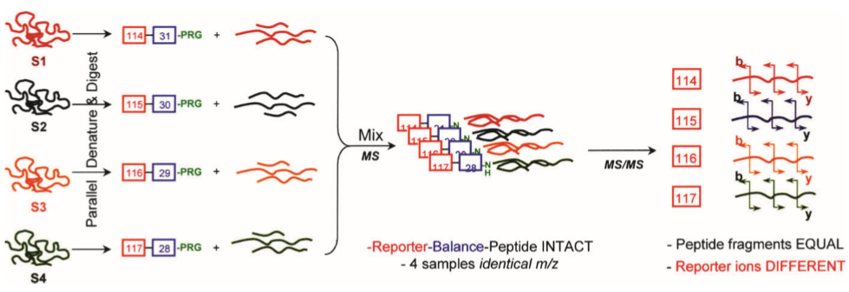
Figure 1. General scheme of a multiplex reaction of four different samples (S1–S4), designated by four different colours (Zieske, et al, 2006)
- The advantages of iTRAQ
1. iTRAQ can simultaneously mark 4 to 8 samples in one experiment. The operation is simple and fast. It is suitable for high-throughput detection of multiple samples, and the experiment design is more flexible.
2. iTRAQ is an in vitro labeling performed at the peptide level and is suitable for many types of biological samples.
3. iTRAQ has the characteristics of good repeatability and high sensitivity, and qualitative and quantitative can be performed simultaneously.
TMT is a peptide in vitro labeling technology developed by Thermo Scientific in the United States. This technology can use up to 10 isotope labels to label the amino groups of polypeptides. After LC-MS / MS analysis, the relative content of proteins in 10 different samples can be compared at the same time. TMT is widely used in the fields of disease marker screening, drug action targets, animal and plant disease resistance or stress resistance mechanisms, animal and plant development and differentiation mechanisms.
- The workflow of TMT
The general process of the TMT is shown in figure 2. Ten different protein samples are denatured, reduced, alkylated and digested into peptide samples. These peptide samples are then labeled with the TMT kit and combined into one sample. Combined samples are separated using HPRP liquid chromatography, and the fractions are concatenated into 12 samples. After purification, each sample is analyzed by HPLC-MS. Data is analyzed to obtain qualitative and relatively quantitative information of the protein.
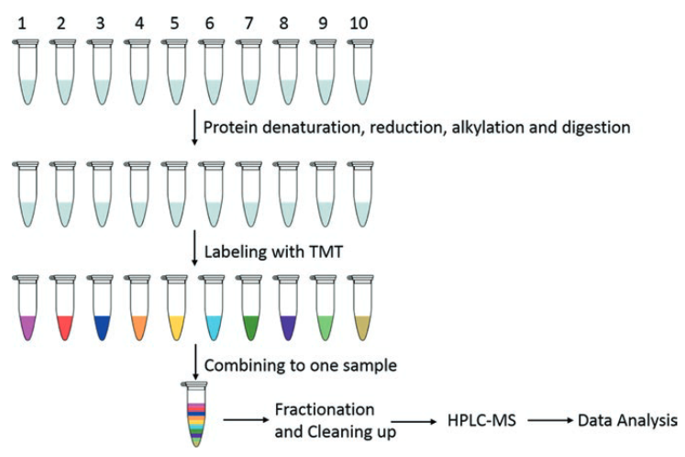
Figure 2. Workflow for relative quantification using 10plex TMT (Zhang, et al, 2017)
- The advantages of TMT
1. High sensitivity: low abundance protein can be detected.
2. Strong separation ability: can separate acid / alkaline protein, protein less than 10KD or greater than 200KD, insoluble protein, etc.
3. Wide scope of application: any type of protein can be identified, including membrane proteins, nuclear proteins, and extracellular proteins.
4. High throughput: 10 samples can be analyzed at the same time, especially suitable for differential protein analysis of samples with multiple processing methods or from multiple processing times.
Stable Isotope Labeling with Amino Acids in Cell culture (SILAC)
SILAC is a powerful approach for high-throughput quantitative proteomics, designed according to the principle of mammalian cell proliferation dependent on essential amino acids. SILAC is applied to quantitative proteome research, which can analyze protein interactions and protein expression differences in cells, providing an effective solution for comprehensive and systematic qualitative and quantitative analysis of complex mammalian cell proteome.
- The workflow of SILAC
The general process of SILAC is shown in figure 3. Adding light, medium or heavy stable isotope-labeled essential amino acids (mainly lysine and arginine) to the cell culture medium, and through the normal metabolism of the cell, the newly synthesized proteins are all tagged with stable isotopes. Mixing all types of protein in equal amounts and after separation by SDS-PAGE, gum cutting, and enzyme digestion, LC-MS / MS analysis can obtain quantitative and qualitative results of related proteins.
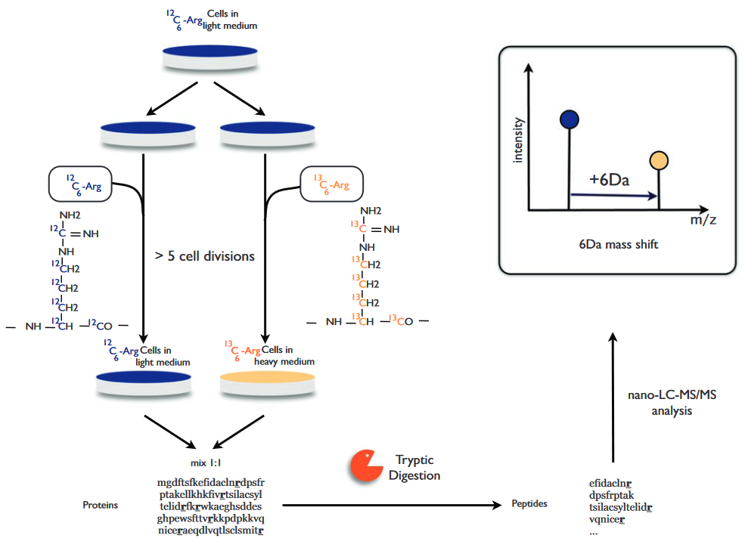
Figure 3. Protein quantification using SILAC (Esthelle et al, 2019).
- The advantages of SILAC
1. SILAC is a living cell-level labeling technology. The labeling effect is stable and efficient, and its labeling efficiency is not affected by lysate.
2. SILAC requires less samples, usually only a few dozen micrograms of protein per sample is sufficient.
3. SILAC uses mass spectrometry to identify and quantify multiple proteins simultaneously.
4. SILAC uses in vivo labeling technology, which is close to the true state of the sample.
References
- Zieske Lynn R. A perspective on the use of iTRAQ™ reagent technology for protein complex and profiling studies. Journal of Experimental Botany, 2006(7):7.
- Lichao Zhang, Joshua E. Elias. Relative Protein Quantification Using Tandem Mass Tag Mass Spectrometry. Methods in Molecular Biology, 2017, 1550:185.
- Esthelle Hoedt, Guoan Zhang, Thomas A. Neubert. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) for Quantitative Proteomics.







