What is Intracellular Protein Crosslinking?
Intracellular protein crosslinking usually refers to the linking of two amino acids between intracellular protein molecules or within protein molecules by covalent bonds other than peptide bonds. Intracellular crosslinking is currently considered a post-translational modification of proteins. It plays an important role in the structure and function of proteins.
It has been found that intracellular protein crosslinks include disulfide bonds, isopeptide bonds, tryptophan crosslinks, tyrosine crosslinks, formaldehyde crosslinks, methylglyoxal (MG) crosslinks, NOS bridges, etc. Studies on disulfide bonds at the histological level have shown that disulfide bonds in proteins can be divided into two major categories, structural and functional, with functional disulfide bonds involved in functions such as catalysis and regulation. Similar analytical work is required for other intracellular crosslinks in proteins. The discovery of these intracellular crosslinking sites will contribute to the study of the regulatory functions of proteins, apoptosis, and disease mechanisms.
Mass spectrometry, with its high throughput and high sensitivity, is one of the tools for large-scale identification of intracellular protein crosslinks. A preliminary set of mass spectrometry-based protein cross-linking analysis method has been developed, including sample pretreatment, data acquisition, qualitative analysis, quantitative analysis and bioinformatics analysis.
Intracellular Protein Crosslinking Assay
- Sample pre-treatment
Sample pretreatment refers to the processing of samples before they are sent to mass spectrometry for data collection. The main purpose is to make crosslinks easier to be collected and accurately identified by mass spectrometry, including enzymatic digestion, separation and enrichment, and amino acid blocking. The specific treatments need to be designed according to the characteristics of the crosslinks.
The purpose of enzymatic digestion is to digest the cross-linked proteins into cross-linked peptides and use them for mass spectrometry data acquisition. Cross-linked peptides can be divided into three basic forms: mono-link, loop-link and cross-link.
Due to the specificity of the enzymatic cleavage site and cross-linking site, and the uneven distribution of amino acids in the protein, not all the cross-linked peptides obtained from the enzymatic digestion of complex samples can be obtained in the above three simple cross-linked forms. Therefore, more diverse complex cross-linked forms need to be considered for large-scale cross-linking identification.
The low content and abundance of crosslinked peptides will result in many crosslinks not being collected and identified. Therefore, performing isolation enrichment is also essential in sample pretreatment. Affinity chromatography, strong cation exchange (SCX) and size exclusion chromatography (SEC) are often used in the separation process. The enrichment at the protein level is performed one step before the enzymatic digestion because the proteins have been digested into peptides after the enzymatic digestion. Enrichment of low abundance proteins can also be achieved by removing high peak proteins during data acquisition using methods such as quadrupole and ion trap.
Amino acid blocking is mainly used to avoid breakage and rearrangement of crosslinks during data acquisition. Most protein crosslinks rely on chemical covalent bonds to directly link amino acids on proteins. The formation process of partial crosslinks is reversible. The crosslinks are easily altered by the added reagents and the environment of the system. These altered cross-links, if incorrectly considered as natural cross-links, will lead to subsequent studies in the wrong direction.
- Data acquisition
Cross-linked peptide identification is performed by comparing the theoretical fragmentation ions generated by the candidate peptide with the actual fragmentation ions collected in the spectrum. There are several ways to perform peptide fragmentation. Different fragmentation methods will result in different fragment ions from the peptide.
Partial cross-linking is the direct joining of two amino acid side chains by chemical bonds, and the fragmentation process breaks the chemical bonds in the peptide segment. Therefore, whether the cross-linked portion is broken during fragmentation requires experiments to demonstrate. Usually, the synthesized standard peptide fragments are subjected to cross-linking experiments, and then they are subjected to data acquisition using mass spectrometry, and the pattern of fragment ions during fragmentation is probed according to the fragment ions in the spectra.
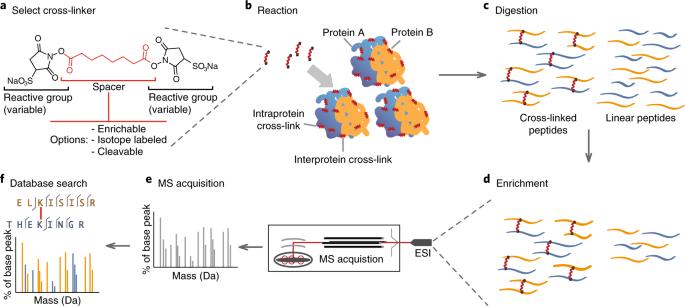
General CLMS workflow (Reilly et al., 2018)
- Qualitative analysis
Theoretical enzymatic cleavage of protein sequences in the database is performed to obtain candidate peptides, and theoretical spectra are generated to match with actual spectra for peptide spectra thus enabling qualitative analysis of protein crosslinks. Currently, there are three main types of cross-linked peptide identification strategies as follows.
Based on unlabeled non-fragmentable cross-linking, the cross-linking does not change throughout the mass spectrum data acquisition process. This type of software cannot determine if the spectrum is that of a cross-linked peptide at the time of identification, so all spectra need to be matched to candidate peptides. This type of crosslinking requires fragmentation of the peptide before acquiring the secondary spectra, but the fragmentation will only occur on the peptide bonds. Thus, the acquired secondary spectrum will have all fragment ions of both peptides under ideal conditions, with some of the fragment ions attached to the other peptide. For this type of identification a search in the space of O(n2) is required.
Unbreakable cross-linking based on isotope labeling. Since crosslinking is the joining of two peptides, the introduction of light and heavy isotopic labeling on the crosslink can be considered to narrow the search. Under the effect of isotopic labeling, two clusters of peaks will appear on the primary spectrum after crosslinking occurs. These two clusters of peaks will meet the mass difference of the light and heavy isotope labeled cross-linker, and thus the spectrum containing the cross-linked peptide can be determined. Since crosslinks labeled with different isotopes have a mass difference, the fragment ions on the secondary spectrum also have the same mass relationship.
Based on the fracturable crosslink, the fracture of the crosslink can be achieved by adjusting the fracture mode and fracture energy. The presence of two fracturable sites on the crosslink will be able to produce four cases in combination with each of the two peptides. This will result in two sets of peaks on the spectrum, each set including two peaks. The difference between these two peaks is the same and is equal to the mass between the two break sites of the cross-linker. Therefore, only these two sets of peaks are searched for during identification to determine the mass of the two peptides that make up the cross-linked peptide, greatly narrowing the search range. This search range is then used to match the fragment ions of the peptides to obtain the optimal results.
- Quantitative analysis
The quantitative analysis of cross-linking is mainly performed on cross-linked peptides and includes labeled and label-free quantification. In labeled quantification, the cross-linked content is reflected by the intensity of the reporter ion. In label-free quantification, the parent ion chromatographic curve corresponding to the cross-linked peptide is reconstructed based on the qualitative results of protein cross-linking. The peak area enclosed by the chromatographic curve reflects the protein cross-linking content.
- Bioinformatics analysis
Bioinformatics analysis can elucidate the possible relationship between crosslinks and protein structure and function. The cross-linked sites are usually mapped to protein sequences based on the identified results, and the protein cross-linkage network is mapped to reflect the distribution of cross-linkages in the protein. The identified cross-linked peptide sites can then be analyzed by motif analysis to identify the hotspots of cross-link formation. The crosslinking of proteins with significant differences may be a potential factor leading to altered protein function and may be an important biomarker.
Reference
- Leitner, A., Faini, M., Stengel, F., & Aebersold, R. (2016). Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends in biochemical sciences, 41(1), 20-32.
- O'Reilly, F. J., & Rappsilber, J. (2018). Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nature structural & molecular biology, 25(11), 1000-1008.






