What Is Post-Translational Modification of Proteins?
Post-translational modification of proteins refers to a covalent process that a protein undergoes during or after translation, that is, changing the properties of a protein by adding one or several amino acid residues to a modification group or by proteolytically cutting the group, which is an important step in the expression of protein biological functions. Post-translational modification of biological proteins greatly increases the types and functions of protein structures. This process regulates the cell cycle and protein transcription, and affect epigenetics.
The Types of Post-Translational Modification
The types of post-translational modification include the removal of N-terminal fMet or Met, the formation of disulfide bonds, chemical modification, and the cleaving of peptide bonds, with phosphorylation being the most common. The chemical modification reaction of proteins is based on maintaining the integrity and function of proteins, and obtaining new bioconjugates through chemical reactions based on their amino acid residues.
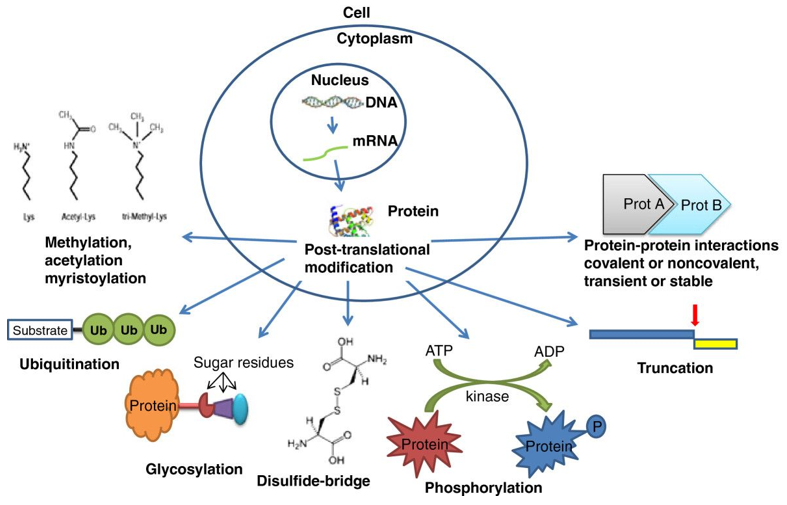
Most common protein PTMs possibly relevant in ASD (Woods et al, 2013).
Where Do Post-Translational Modifications Occur?
Post-translational modifications mainly occur at the side chains of amino acids or at the C or N terminus of proteins. The existing functional groups or the introduction of new functional groups can be used to modify proteins to extend the chemical composition of 20 standard amino acids. The sites for post-translational modification are often with functional groups that can act as nucleophiles in chemical reactions, such as the hydroxyl groups of serine, threonine, and tyrosine; lysine, arginine, and histidine amine form; N and C terminus of the protein, at the same time, asparagine amide can also be used as a kind of glycan connection point in post-translational modifications. Post-translational modifications also occur on oxidized methionine and some methylene groups in the side chain site.
Research Strategies of Post-Translation Modification
At present, there are several strategies to analyze protein post-translational modifications, including mass spectrometry (MS), Eastern blotting, radioactive labeling, and Western blotting. Radioactive labeling and Western blotting are the most traditional, specific and relatively quantitative. The process requires prior knowledge of post-translational modification types. The limitations inlcude antibody specificity and availability. Mass spectrometry is also commonly used to provide a general information on the modification. It is not limited by the prior knowledge on the type of the modification and related modification sites.
- Bottom-up
Bottom-up technology is one of the mass spectrometry techniques used in proteomics research. It requires the use of proteases to digest the protein sequence before identifying the peptide based on the sequence. Since this technique cannot guarantee the integrity and stability of the detected peptides during the detection process, information about the relationship between post-translational modifications of different proteins cannot be obtained, which means that it is unable to detect certain modifications. Therefore, this method is not suitable for analyzing the combined post-translational modified code of histones.
- Middle-down
The middle-down analysis method is an alternative to the bottom-up analysis method, and its principle is similar to that of the bottom-up technology when analyzing histones. When using this analysis method, the detected protein usually needs to be digested into peptides within the range of 3-9 kDa, so that the integrity of the detected peptide cannot be guaranteed. But the middle-down analysis method is gradually gaining popularity, mainly due to the advancement in the instrument and the combined modification that preserved histone tails. The middle-down is closer to the sensitivity of the bottom-up method.
Top-down technology can directly sequence intact proteins, including post-translational modified proteins and other large protein fragments, not just peptides. This allows PTMs-related information to be preserved to the maximum extent to make it suitable for the comprehensive characterization and analysis of the histone code. The effective resolution of the top-down technology for proteins has reached 229kDa. The number of proteins that can be detected at a time is also increasing. At present, Top-down technology is mainly used to analyze single protein, and high-throughput analysis technology has not been widely used.
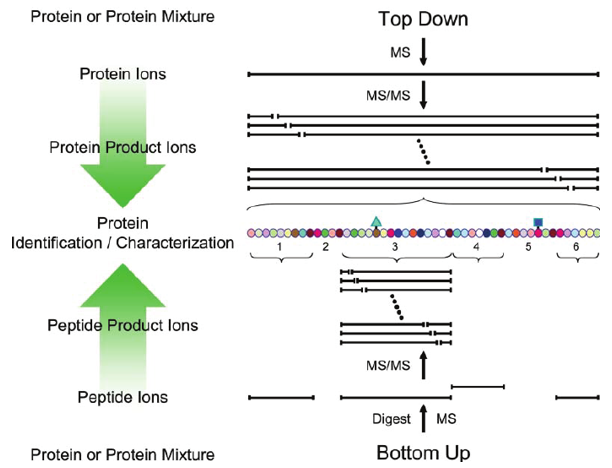
Schematic overview of bottom-up and top-down approaches employed for tandem MS-based protein identification and characterization (Scherperel et al, 2007).
References
- Wang Y C, Peterson S E, Loring J F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell research, 2014, 24(2): 143.
- Silva, André MN, et al. "Post-translational modifications and mass spectrometry detection." Free radical biology and medicine, 2013(65): 925-941.
- Drazic A, Myklebust L M, Ree R, et al. The world of protein acetylation. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 2016, 1864(10): 1372-1401.
- Woods, A.G., Ngounou Wetie, A.G., Sokolowska, I. et al. Mass spectrometry as a tool for studying autism spectrum disorder. J Mol Psychiatr, 2013, (1): 6.
- Scherperel G, Reid G E. Emerging methods in proteomics: top-down protein characterization by multistage tandem mass spectrometry. Analyst, 2007, 132.






