Molecular Diversity and Structural Geometry
Biophysical Dynamics of the Lipid Bilayer
Membrane Asymmetry and Domain Organization
Protein-Lipid Interactions and Anchoring Mechanisms
Technical Sovereignty: Characterizing Membrane States in Research Models
Conclusion
FAQ
A membrane is often introduced as a barrier. That definition is correct, but it is too weak for serious membrane biology. A living membrane is not just a wall between two aqueous compartments. It is a mechanically responsive surface. It stores elastic stress. It controls local charge. It redistributes molecules laterally. It bends, buds, and fuses without losing continuity. It also creates selective landing zones for proteins, lipids, and ions. Once that is understood, phospholipids stop looking like generic building blocks and start looking like the structural language of membrane behavior.
That language is subtle. Small changes in head-group area, acyl-chain saturation, leaflet placement, or local ionic environment can shift the state of the whole membrane. A phosphatidylcholine-rich surface does not behave like a phosphatidylethanolamine-rich one. A phosphatidylserine-enriched inner leaflet does not present the same electrostatic field as an outer leaflet dominated by phosphatidylcholine and sphingomyelin. A small phosphoinositide pool can reorganize a membrane zone without causing a dramatic change in total phospholipid mass. In other words, membrane function does not follow from lipid presence alone. It follows from lipid geometry, distribution, and collective behavior.
This is exactly where many phospholipid articles become too generic. They explain the hydrophilic head and hydrophobic tails, then jump to a vague statement about "maintaining membrane fluidity." That summary is not wrong, but it misses the real problem. The more useful questions are harder. Why do some lipids stabilize flat lamellar surfaces while others lower the energetic penalty of high curvature? Why is lateral diffusion rapid while spontaneous flip-flop remains slow? Why do ordered domains emerge only in certain compositional windows? Why can protein recruitment depend as much on charge shielding and head-group spacing as on the presence of one named lipid species?
In membrane research, the most common mistake is not missing a lipid class. It is choosing a readout that cannot answer the membrane question being asked. A bulk composition assay cannot directly resolve leaflet asymmetry. A mobility assay cannot tell you which phospholipid species changed. A fluorescent membrane dye does not automatically report order, hydration, and phase state at the same time. That is why membrane biophysics and structural phospholipidomics work best when composition, phase behavior, curvature logic, and assay choice are kept in one frame. A project may begin with a broad lipidomics profiling workflow, but it becomes far more useful when the membrane question is narrowed to geometry, asymmetry, or signaling-active lipid pools.
Molecular Diversity and Structural Geometry
Phospholipid classes are physical instructions, not just chemical names
Glycerophospholipids are often presented as a neat family built on one shared scaffold: glycerol, two hydrophobic chains, and a phosphate-linked head group. That is chemically true, but physically incomplete. From the membrane's perspective, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, and phosphatidylglycerol are not minor variations on one theme. They are different solutions to the same design problem: how to organize a deformable interface between water and a hydrophobic core.
Phosphatidylcholine (PC) is the classic lamellar phospholipid. Its choline head group is relatively bulky, so the effective interfacial area remains broad. In many mixtures, this gives PC a more cylindrical shape. Cylindrical lipids stabilize flat bilayers because the head-group area and hydrophobic cross-section are relatively well matched. That makes PC especially useful for maintaining broad bilayer continuity.
Phosphatidylethanolamine (PE) shifts that balance. Its head group is smaller than choline, so the interfacial area contracts relative to the hydrophobic region. The molecule becomes more cone-like. That change has large consequences. Cone-shaped lipids are less comfortable in perfectly flat bilayers and more compatible with negative curvature. A PE-rich membrane can remain bilayered, but it stores a stronger preference for curvature. That preference becomes important when the membrane forms budding necks, fusion intermediates, or other highly deformed states.
Phosphatidylserine (PS) introduces a different variable. It does not mainly change curvature logic. It changes surface electrostatics. A PS-rich leaflet presents a more anionic interface, which alters ion association, protein recruitment, and local membrane potential. In many cells, that is one reason PS is so valuable on the cytosolic side of the plasma membrane.
Phosphatidylinositol (PI) is even more strategically important because its inositol head group can be phosphorylated into multiple signaling-active forms. PI is therefore not just another anionic phospholipid. It is a platform for membrane coding. Small phosphoinositide pools can define membrane identity, recruit specialized domains, and control trafficking decisions without requiring a large change in the total phospholipid pool. When that signaling layer becomes the central mechanistic research question, a focused targeted phosphoinositide-resolved analysis is often more informative than a broad phospholipid total.
Phosphatidylglycerol (PG) matters strongly in bacterial membranes and in organelle-linked lipid metabolism. The broader lesson is simple. These classes should not be treated as a taxonomic checklist. Each one changes the membrane's physical problem in a different way. Some favor flatness. Some favor curvature. Some reshape charge. Some influence sterol compatibility. Some alter head-group spacing and hydration. The membrane reads all of those variables together.
That is why total phospholipid abundance is often too blunt to explain membrane behavior. Two membranes may contain similar total phospholipid mass and still behave very differently if one is enriched in PC and sphingomyelin while the other contains more PE, PS, and PI. The first is more likely to support tighter packing and more ordered lateral organization. The second is more likely to present a more negative surface, stronger curvature bias, and a different protein-recruitment landscape. When this level of architecture matters, dedicated glycerophospholipid profiling or a focused phospholipid class-and-species analysis is usually more informative than a single aggregate readout.
Critical packing parameter turns chemistry into membrane prediction
A useful way to formalize this logic is the critical packing parameter (CPP), written as $v / (a \cdot l_c)$, where $v$ is hydrophobic volume, $a$ is effective head-group area, and $l_c$ is chain length. The formula itself is compact. Its value is conceptual. It explains why certain lipids prefer flat lamellae, while others are more compatible with highly curved or non-lamellar states.
When head-group area and hydrophobic volume are balanced in a more cylindrical way, bilayers are favored. When the head-group area becomes relatively small, the molecule becomes more conical, and the system begins to favor negative curvature. As this preference strengthens, non-lamellar structures become easier to access. The membrane is no longer just a pile of amphiphiles. It becomes a geometry-driven self-assembly system.
This is where phospholipid structure becomes predictive. PC usually lies closer to the lamellar end of the continuum. PE shifts the system toward stronger curvature stress. Phosphatidic acid can become highly curvature-active because its effective head-group behavior is sensitive to protonation and local ion concentration. Small structural differences therefore produce large topological consequences.
The most important point is that non-lamellar propensity matters even when the membrane stays bilayered. A nominally lamellar membrane can still contain lipids whose preferred geometry is not flat. Those lipids store curvature stress. They reduce the energy barrier for intermediate states. They help explain why some membranes bud more easily, why some fusion events are less costly, and why some remodeling proteins function only in specific lipid environments.
Lipid polymorphism is central, not optional
Membrane discussions often stop at the bilayer, but phospholipid systems are polymorphic. Depending on composition, temperature, hydration, and ionic environment, they can adopt multiple mesophases. The fluid lamellar phase, $L_{\alpha}$, is the familiar bilayer state. The inverted hexagonal phase, $H_{II}$, reflects a strong preference for negative curvature. Bicontinuous cubic phases, often grouped as $Q_{II}$, represent highly curved, interconnected membrane geometries.
The biological importance of this is often misunderstood. It is not that living cells routinely convert whole membranes into stable hexagonal or cubic phases. The real importance is that these tendencies reveal what kinds of curvature and remodeling a composition can tolerate. A membrane enriched in lipids with strong non-lamellar preference will not respond to budding, fusion, or scission the same way as a membrane dominated by lamellar-stabilizing species.
This is also why membrane remodeling is never only a protein story. Proteins provide force, scaffolding, or local specificity, but the lipid matrix determines how expensive a shape change will be. If the bilayer already stores the right curvature preference, the protein is working on a permissive material. If the membrane strongly resists that geometry, the same protein action costs more.
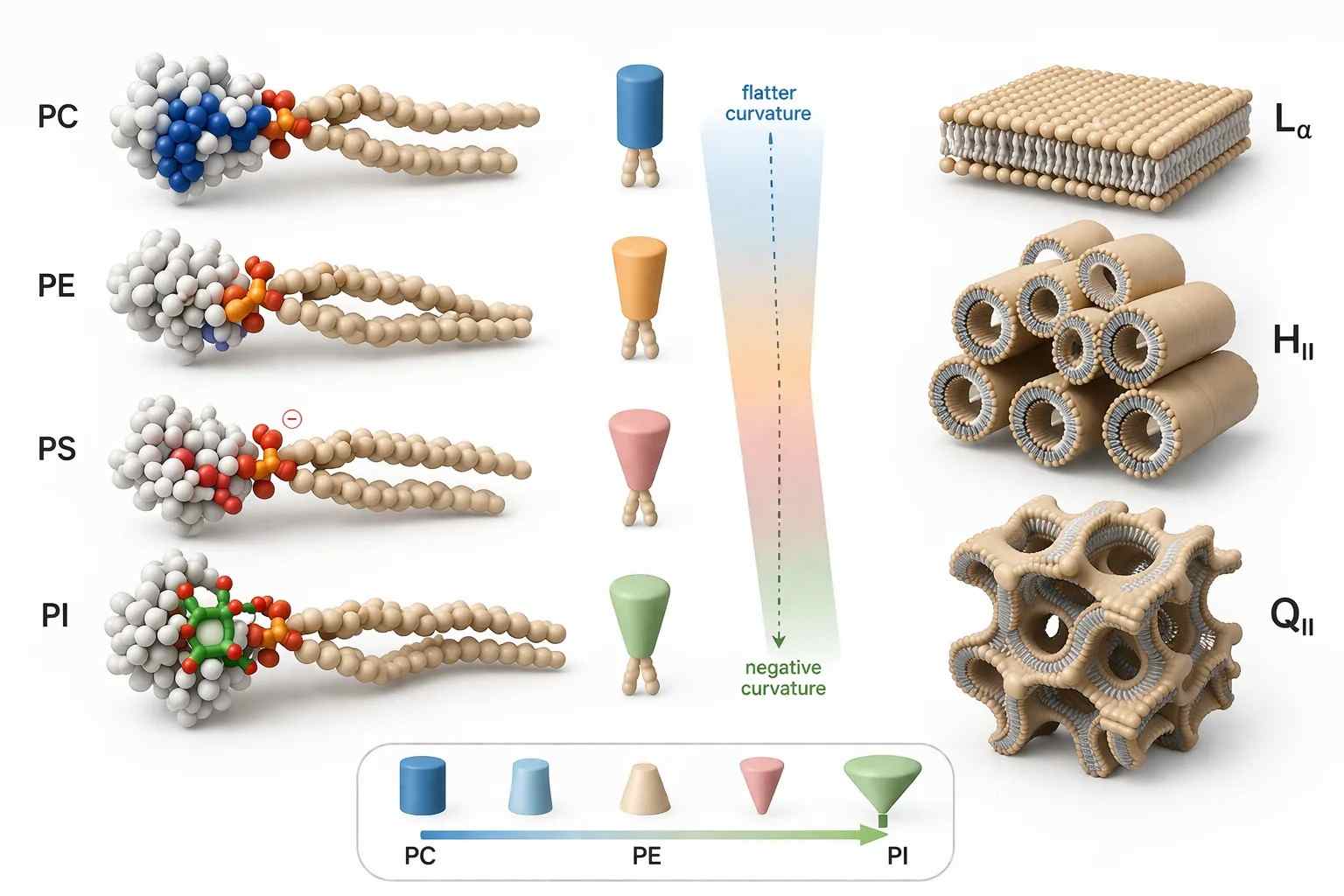
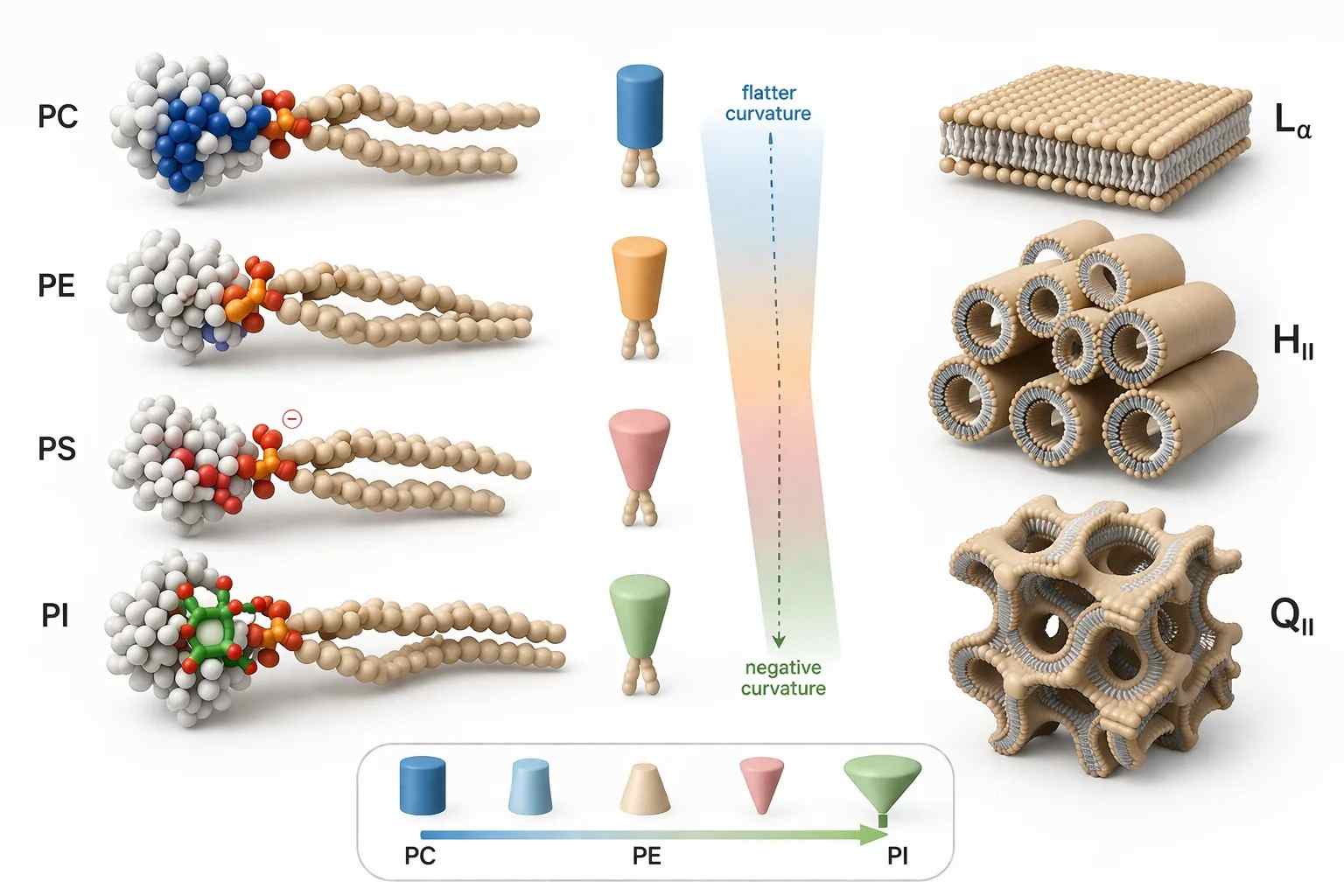 Figure 1. Phospholipid geometry and phase preference.
Figure 1. Phospholipid geometry and phase preference.
What it shows: Cylindrical and cone-shaped phospholipid classes arranged against CPP-guided transitions toward lamellar ($L_{\alpha}$), inverted hexagonal ($H_{II}$), and bicontinuous cubic ($Q_{II}$)-like membrane states.
The figure belongs here because this is the point where the argument shifts from chemical class to membrane topology. Once geometry is visualized, the link between phospholipid structure and self-assembly becomes much easier to read.
Head groups and acyl chains must be interpreted together
A common analytical mistake is to separate head-group logic from acyl-chain logic too sharply. In real membranes, the two are coupled. The head group defines the interfacial problem. The hydrophobic chains define the packing and thermodynamic problem. The membrane experiences both at once.
Take PC as an example. The class label alone does not tell the full story. A PC species carrying two saturated chains behaves differently from a PC species carrying one or more cis-unsaturated chains. The head group may still support a broad lamellar outline, but the chains determine how tightly the membrane packs, how thick it becomes, how close it sits to a phase boundary, and how compatible it is with ordered domains.
The same principle applies to PS and PI. Their anionic head groups shape the electrostatic interface, but chain composition still determines the physical background on which that electrostatic effect is expressed. A polyunsaturated PS species does not create the same local packing environment as a more saturated PS species. A signaling-rich phosphoinositide patch embedded in a tightly packed membrane does not behave the same way as the same lipid pool embedded in a softer, defect-rich membrane.
This is why species-level resolution matters so much. A membrane can appear stable at the class level while undergoing meaningful reorganization at the molecular-species level. A shift from saturated C16/C18 species toward longer or more unsaturated species may significantly alter phase behavior, permeability, curvature tolerance, and protein partitioning even if the class distribution looks only modestly different. In studies that need to move beyond class totals, a targeted species-level lipid workflow often provides more mechanistic traction than broad totals alone, while a broad discovery lipid survey remains useful when the first goal is to map the wider phospholipid landscape before narrowing the mechanism.
Biophysical Dynamics of the Lipid Bilayer
Phase transitions define membrane state more precisely than "fluidity"
"Membrane fluidity" is convenient language, but it is too vague for advanced analysis. A membrane is not simply more fluid or less fluid. It occupies composition-dependent phase states. The classical transition is between the gel phase, $L_{\beta}$, and the liquid-crystalline phase, $L_{\alpha}$.
In the gel phase, acyl chains are more ordered and extended. Free volume is lower. Lateral mobility is reduced. The membrane is usually thicker and less deformable. In the liquid-crystalline phase, chain disorder rises, lateral diffusion becomes much easier, and the bilayer becomes softer and more dynamic. The transition temperature, $T_m$, marks where this shift becomes prominent for a given composition.
This matters because membrane behavior can change sharply near a phase boundary. A small shift in temperature, sterol content, chain length, or unsaturation can alter thickness, permeability, mechanical compliance, and probe response. A membrane near transition is not simply "partly fluid." It is physically sensitive. That sensitivity makes otherwise modest compositional changes much more consequential.
For experimental design, this point is fundamental. A membrane model built without regard to phase position may appear to respond to a biological variable when the real driver is hidden thermodynamic placement. The same nominal composition can behave differently at different temperatures or hydration conditions if it sits close to a transition regime.
Chain length and unsaturation are the dominant thermodynamic levers
Among the many variables that shape membrane state, acyl-chain length is one of the most direct. Longer chains create more van der Waals contact and usually favor tighter packing. That pushes the system toward higher order and often raises $T_m$. Shorter chains reduce those interactions, which tends to lower $T_m$ and increase disorder.
Unsaturation acts just as strongly, but in the opposite structural direction. Cis double bonds introduce kinks into the hydrophobic chain. Those kinks disrupt tight chain alignment and make highly ordered packing harder to maintain. The result is lower $T_m$, increased chain disorder, greater free volume, and often greater membrane deformability.
A membrane enriched in unsaturated species is therefore not merely "more fluid." It is physically different in several connected ways. It is more permissive to transient packing defects. It often responds differently to cholesterol. It may also alter the ease with which amphipathic helices or shallow insertion motifs engage the bilayer. This is why the contrast between saturated and unsaturated species has such strong consequences. A membrane enriched in saturated C18 chains may remain relatively ordered under conditions where a membrane rich in cis-unsaturated C18 species is already highly dynamic.
Lateral diffusion is fast, but flip-flop is not
One of the defining features of membranes is the contrast between in-plane motion and transbilayer motion. Within one leaflet, many lipids move laterally with surprising speed. That mobility allows a membrane to reorganize around a local event. A protein can enrich its preferred lipid environment. A signaling site can gather selected components. A budding region can become compositionally distinct without requiring the whole membrane to change at once.
Across the bilayer, the story is different. A phospholipid head group is polar or charged, so moving it through the hydrophobic core is energetically expensive. That is why spontaneous flip-flop is slow for many phospholipids. In protein-free systems, redistribution across leaflets often occurs on timescales of hours rather than seconds.
This kinetic contrast is one of the hidden foundations of membrane asymmetry. If flip-flop were intrinsically fast, biologically useful leaflet differences would decay much more easily. Instead, the two sides of the membrane can preserve distinct chemical identities. The inner leaflet can remain enriched in aminophospholipids and phosphoinositides, while the outer leaflet maintains a different interfacial character.
Flippases, floppases, and scramblases make asymmetry a maintained state
Leaflet asymmetry persists because cells actively manage it. Flippases, floppases, and scramblases do not perform the same task, but together they determine whether lipids remain segregated, are selectively transferred, or become rapidly redistributed across the bilayer. This means membrane asymmetry is not a passive resting pattern. It is an energy-dependent and regulated property.
That insight changes how membrane experiments should be interpreted. A shift in surface PS exposure does not always mean total PS abundance changed. A signaling event can alter lipid presentation by transport rather than synthesis. Likewise, a model membrane that lacks these transport systems may reproduce phase behavior well while failing to preserve physiological leaflet asymmetry over time. When the goal is to connect lipid remodeling with compartment-resolved biology, phospholipid profiling often becomes more informative when paired with research-use-only subcellular proteomics workflows.
Membranes are mixtures under competing structural instructions
No biological membrane is built from one lipid species. Saturated chains push toward order. Unsaturated chains push toward disorder. Bulky head groups favor lamellar packing. Smaller head groups favor curvature. Cholesterol condenses some backgrounds and loosens others. Anionic lipids alter surface potential and ion sensitivity. The actual membrane state emerges from the compromise among these competing tendencies.
This is where many experimental misreadings begin. A single probe signal may be overread as "higher order" when the real shift is altered hydration or sterol redistribution. A class-level lipid increase may look decisive when the real change lies in species balance. A mobility change may be attributed to composition when the stronger driver is protein crowding or domain confinement. Good membrane analysis therefore relies on triangulation between composition and state. In practice, that often means pairing membrane-state measurements with pathway-level interpretation of lipid remodeling so that observed physical shifts can be traced back to plausible compositional drivers.
Membrane Asymmetry and Domain Organization
The outer leaflet and inner leaflet are not equivalent surfaces
A biological membrane is not a mirror-symmetric sandwich. The exoplasmic and cytosolic leaflets present different chemical faces, and that difference is one of the reasons membranes can support directional signaling and selective recruitment. In many plasma membranes, sphingomyelin and phosphatidylcholine are enriched on the outer side, while phosphatidylethanolamine, phosphatidylserine, and most phosphoinositides are concentrated on the inner side.
The consequence is larger than composition alone. A PS-rich cytosolic leaflet presents a different electrostatic field from a neutral outer leaflet. A PE-enriched inner side stores a different curvature bias. A phosphoinositide-rich surface can selectively recruit specific protein modules even when the total membrane composition appears relatively stable. That is why bulk phospholipid totals cannot fully answer a leaflet-specific membrane question.
Liquid-ordered domains are physical states before they are signaling platforms
The term "lipid raft" has often been oversimplified. A more useful view is that certain membrane mixtures can laterally separate into regions with different physical states. Cholesterol and sphingomyelin are especially important because they can pack together in a way that stabilizes the liquid-ordered, or $L_o$, state. That state is more ordered than the surrounding membrane but still retains lateral mobility.
This distinction matters because proteins do not experience the membrane as a chemically uniform sheet. An ordered patch can differ in thickness, packing density, compressibility, and partitioning preference. That means membrane heterogeneity is not only compositional. It is physical. A signaling platform is often the consequence of a favorable membrane state, not just a named lipid list.
Old diagrams often depict rafts as large, stable islands floating in a fluid sea. That picture is too rigid. A more accurate view is dynamic, nanoscale, and composition-dependent. Domain contrast matters, but it should not be drawn as a permanent cartoon island detached from the rest of the membrane.
Curvature hotspots are compositionally biased
Highly curved membrane regions such as endocytic necks, budding rims, and remodeling sites impose a strong energetic penalty. Because of that, they are rarely compositionally neutral. Lipids such as PE and phosphatidic acid become especially important because they lower the cost of negative curvature or curvature stress.
This does not mean a budding neck contains only curvature-active lipids. Real membranes remain mixed. The more useful point is that some lipids make extreme topology easier and others resist it. A membrane preparing for vesicle formation or scission will therefore not behave as a random slice of the average bilayer.
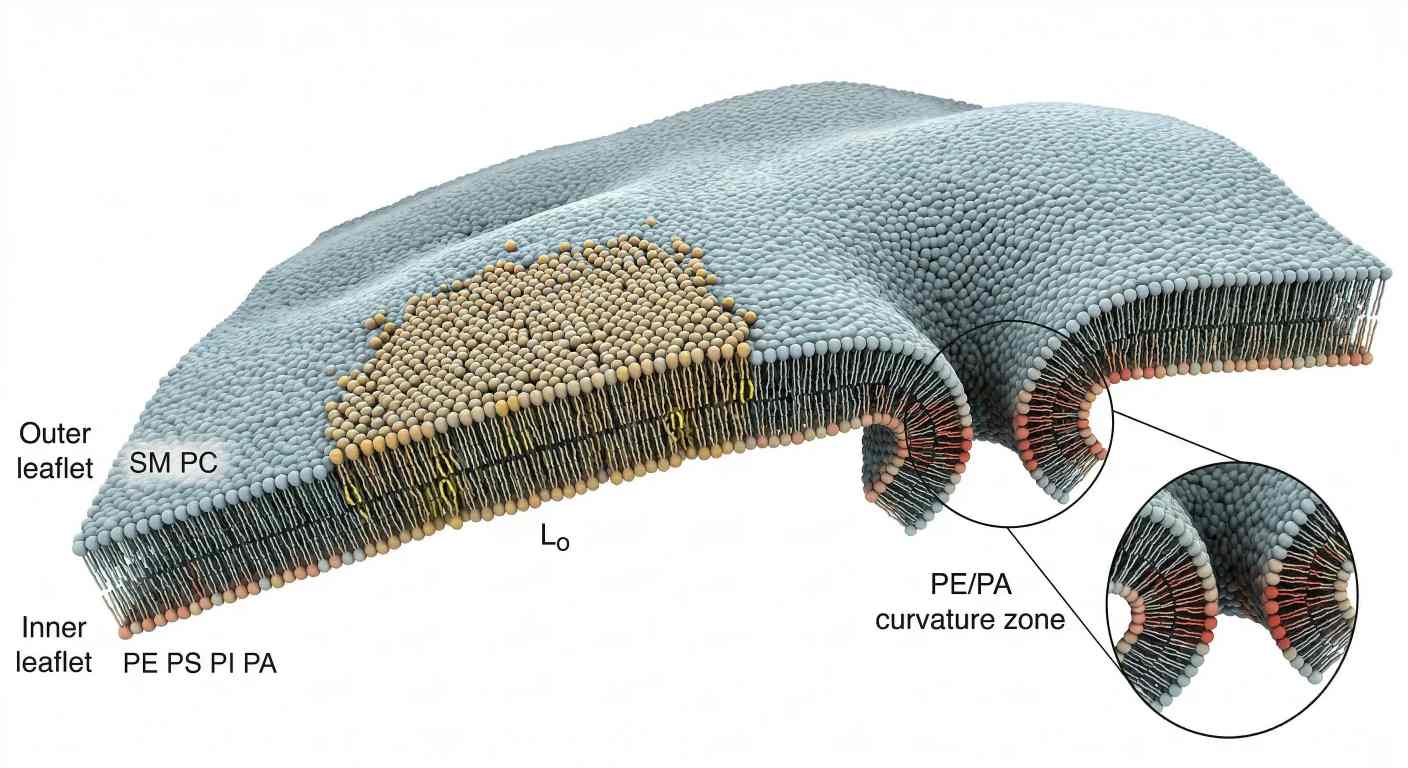 Figure 2. Membrane asymmetry, ordered domains, and curvature hotspots.
Figure 2. Membrane asymmetry, ordered domains, and curvature hotspots.
What it shows: A cross-sectional membrane view integrating outer-versus-inner leaflet asymmetry, a cholesterol- and sphingomyelin-enriched liquid-ordered patch, and a high-curvature region biased toward PE- and PA-rich remodeling.
The second figure belongs here because this is the point where the article shifts from molecular and thermodynamic logic into spatial organization. Readers need to see leaflet asymmetry, ordered domain formation, and curvature bias in one integrated membrane landscape.
Protein-Lipid Interactions and Anchoring Mechanisms
Membrane recruitment begins with surface chemistry
Proteins do not bind "the membrane" in the abstract. They bind a specific interfacial environment. That environment is defined by charge density, head-group spacing, hydration, lateral order, and local curvature. A membrane patch enriched in phosphatidylserine does not present the same binding surface as one dominated by phosphatidylcholine. A phosphoinositide-rich zone is even more selective because it combines negative charge with head-group-specific recognition.
This is why protein recruitment cannot be explained by lipid identity alone. The same phospholipid class can support different binding outcomes depending on chain order, sterol content, ionic strength, and membrane geometry. A protein approaching the membrane is therefore not reading one variable. It is reading a composite physical code.
That point matters especially in crowded signaling environments. Some proteins need a broadly anionic surface. Some need one specific phosphoinositide. Some need both. Others require the membrane to expose shallow packing defects so that an amphipathic segment can insert. In practice, the membrane decides which mode of binding is energetically favored.
Anionic lipids create selective recruitment platforms
Phosphatidylserine and phosphoinositides are especially important because they reshape the electrostatic field at the membrane surface. A PS-rich cytosolic leaflet creates a negative interface that can attract polybasic regions on peripheral proteins. Phosphoinositides add another layer of selectivity because their phosphorylated inositol head groups can be recognized by dedicated binding domains.
This means the membrane is not only a site of recruitment. It is also a filter. Two proteins may both carry basic residues, yet only one may bind stably if one requires a defined phosphoinositide signature and the other only senses general surface charge. The result is spatial sorting at the nanometer scale.
When a project needs to move from bulk membrane composition to signaling-active phospholipid pools, targeted phosphoinositide-resolved analysis is often more informative than a class-total measurement, because the biological question is no longer "how much phospholipid is present" but "which membrane code is actually exposed."
PH domains and C2 domains read different parts of the membrane code
Pleckstrin homology domains are often used as the classic example of phosphoinositide-sensitive recruitment. That is useful, but it should not be simplified into the claim that all PH domains behave the same way. What matters is that many of them recognize defined phosphoinositide environments and therefore localize proteins to specific membrane regions.
C2 domains illustrate a different logic. They often couple membrane association to calcium-dependent or calcium-assisted interactions with anionic lipids. That means membrane binding can become highly conditional. A protein may have the right domain architecture and still remain weakly membrane-associated until local divalent cation conditions shift.
These examples show why membrane recruitment is combinatorial. One protein may need a phosphoinositide signature plus general negative charge. Another may need calcium plus PS exposure. Another may need electrostatic attraction plus favorable curvature. The membrane becomes a selective landing field because it presents multiple variables at once.
When the question shifts from membrane composition to the protein layer that responds to that composition, membrane protein identification support can help connect lipid-defined surfaces with the proteins most likely to partition into or respond to them.
Hydrophobic insertion is a separate energetic problem
Not all membrane association is purely electrostatic. Many proteins also use shallow hydrophobic insertion. An amphipathic helix is the clearest example. One face of the helix presents hydrophobic residues that can insert into the upper hydrophobic region of the bilayer, while the opposite face remains compatible with water or charged head groups.
This kind of binding is strongly affected by packing defects. A tightly packed, ordered membrane can resist shallow insertion more strongly than a disordered or curvature-stressed membrane. By contrast, an unsaturated or highly curved membrane often exposes transient defects that make insertion easier. The membrane is therefore not only deciding whether a charged domain can approach. It is also deciding whether the physical structure will accept penetration.
This distinction matters because some proteins prefer the body of an ordered domain, while others prefer its edge. Some partition into defect-rich disordered regions. Others need curvature-induced packing stress before they bind efficiently. A membrane-binding event is often best understood as a negotiation between electrostatics and insertion energetics.
GPI anchoring solves localization in a different way
GPI anchoring follows a different logic from reversible peripheral recruitment. Here the protein is covalently linked to a glycolipid anchor that inserts into the membrane. The protein is therefore pre-positioned at the exoplasmic leaflet.
This does not remove lipid dependence. It changes the form of that dependence. GPI-anchored proteins still sort according to local membrane state, sterol compatibility, and trafficking route. They can accumulate differently in ordered or disordered regions, and their surface distribution remains sensitive to the lipid background. Covalent attachment solves one localization problem, but it does not erase membrane physics.
Local pH and ion concentration can reweight binding affinity
Local chemistry can shift membrane binding even when lipid composition remains unchanged. Divalent cations such as Ca²⁺ and Mg²⁺ can partially shield anionic head groups, reduce repulsion between neighboring lipids, and alter the effective surface field seen by proteins. Local pH can also modify protonation-sensitive lipids, especially phosphatidic acid, and thereby change head-group behavior.
This means the same membrane composition can support different recruitment outcomes under different buffer conditions. A PS- and PI-rich membrane may be strongly recruiting in one context and much less so in another if shielding or protonation changes the interfacial charge landscape. This is one reason assay design matters so much. Membrane biophysics is often controlled as much by the test environment as by the nominal lipid recipe.
When membrane-binding effects need to be connected to downstream assemblies rather than isolated interactions, downstream interaction-network analysis can be a useful follow-up because it helps separate primary membrane recruitment from the secondary interaction networks that form afterward.
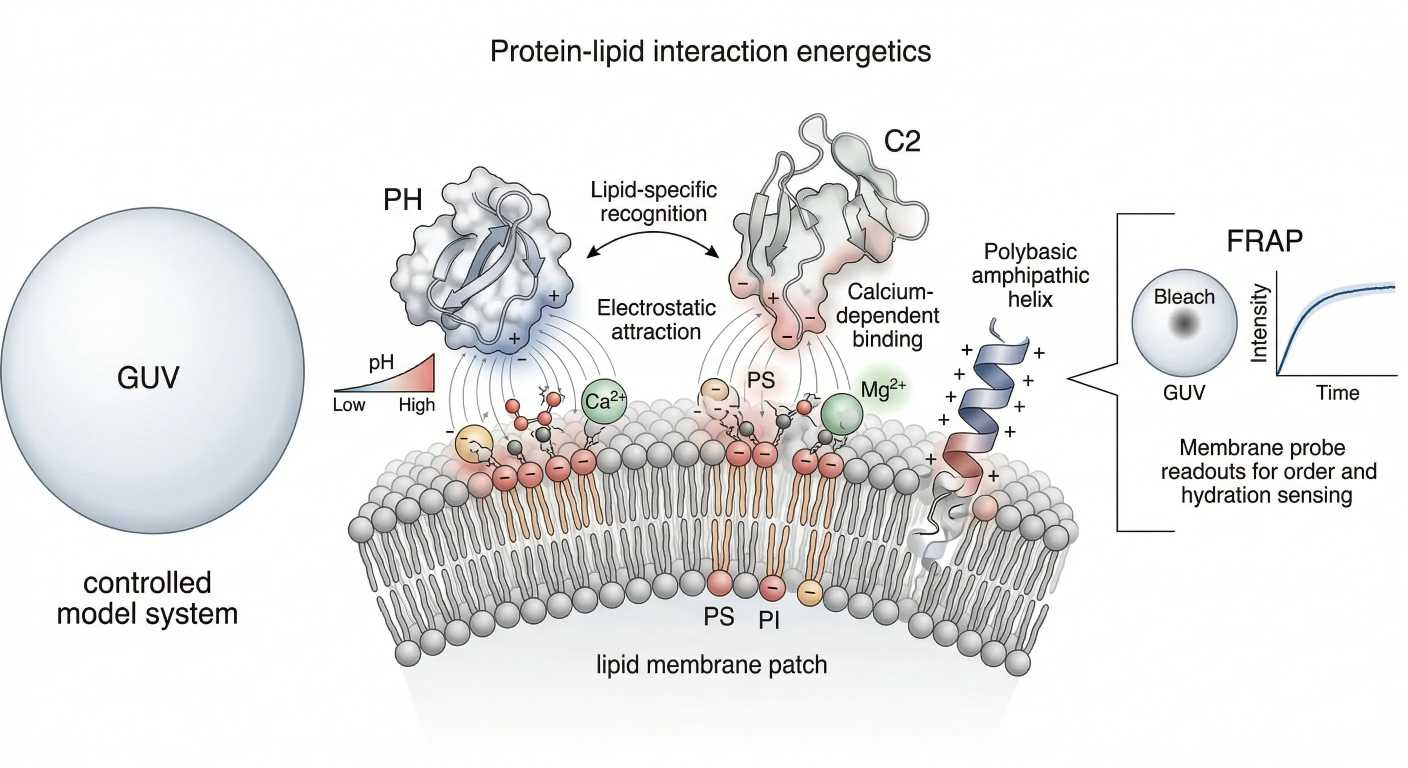 Figure 3. Protein recruitment energetics and membrane-state readouts.
Figure 3. Protein recruitment energetics and membrane-state readouts.
What it shows: An integrated membrane scene combining electrostatic recruitment to PS- and phosphoinositide-rich surfaces, shallow hydrophobic insertion into defect-prone regions, ion shielding by Ca²⁺ or Mg²⁺, and the transition from membrane mechanism to assay logic through GUV-, FRAP-, and probe-based readouts.
The third figure belongs here because this is the point where membrane mechanism and assay design converge. Readers need to see that protein recruitment is not only a biochemical event. It is also a measurable physical event whose outcome depends on surface charge, insertion opportunity, and local chemical context.
Technical Sovereignty: Characterizing Membrane States in Research Models
FRAP measures mobility, not composition
Fluorescence recovery after photobleaching is powerful because it answers one very specific question well: how mobile is the labeled membrane component after a local bleach event? In a fluid membrane, lateral diffusion repopulates the bleached area quickly. In a constrained membrane, recovery is slower or incomplete.
The value of FRAP lies in that specificity. It is a motion readout, not a composition readout. It can tell you that a membrane region is more mobile, more confined, or more heterogeneous. It cannot tell you by itself which phospholipid species created that behavior. This is why FRAP and lipidomics should not be treated as substitutes. They answer different questions.
Used well, FRAP becomes especially informative when the experiment asks whether a membrane region behaves as an open fluid, a scaffolded patch, or a domain-constrained environment. If the membrane question is about motion, FRAP is aligned. If the membrane question is about class composition or species remodeling, FRAP alone is not enough.
GUVs are useful because they remove complexity on purpose
Giant unilamellar vesicles are powerful not because they imitate every feature of a living cell, but because they do not. They remove cytoskeletal coupling, transporter activity, and most trafficking noise. That simplification lets researchers control composition, temperature, phase coexistence, and curvature without too many overlapping variables.
This makes GUVs ideal for questions about intrinsic membrane behavior. Can a given lipid mixture phase separate? Does a protein partition into a curved or disordered region? Does a probe preferentially report interfacial hydration or domain contrast? These are questions that often become clearer in a simplified model than in a living membrane crowded with biological confounders.
That simplification is a strength, not a weakness, as long as the limitation is understood. GUVs are not substitutes for cells. They are controlled test systems for membrane-state logic. In that sense, they provide technical sovereignty: the ability to isolate one membrane variable at a time and observe what it actually does.
Probe choice determines what "membrane order" really means
A common mistake in membrane work is to talk about fluorescent probes as though they all report the same property. They do not. Each probe reports through a different physical mechanism, and that mechanism determines what the data can mean.
Laurdan is especially useful because it is sensitive to interfacial hydration and dipolar relaxation. That makes it valuable for distinguishing membrane regions that differ in water accessibility and packing state. It is often the best choice when the real question is about how the interface changes rather than how the hydrophobic core behaves in isolation.
DPH reports differently. It sits deeper in the membrane and is commonly used to infer core order through anisotropy-related measurements. It is less direct for interfacial hydration and head-group-level phenomena.
DiI and related membrane dyes are useful for visualization and phase preference studies, but they are not universal order sensors. Their partitioning behavior depends on the exact analog and the membrane environment.
The practical lesson is simple. Probe selection should begin with the property of interest. If the question is hydration-sensitive order mapping, Laurdan is often more informative. If the question is core order, DPH may be better aligned. If the goal is imaging membrane partitioning, DiI-like probes may be convenient, but they should not be overinterpreted.
Assay selection by membrane question
| Research question | Best-aligned readout | What it can answer | What it cannot answer |
|---|---|---|---|
| Is this membrane region mobile or confined? | FRAP | Lateral recovery kinetics and immobile fraction | Exact lipid species responsible |
| Can this composition phase-separate or sort by curvature? | GUV-based model membranes | Intrinsic phase behavior, curvature sorting, domain preference | Full physiological transporter or cytoskeletal control |
| Is the interface becoming more hydrated or phase-shifted? | Laurdan-type probes | Hydration-sensitive membrane-state changes | Full molecular composition |
| Is the hydrophobic core becoming more ordered? | DPH-type probes | Changes in core order and rotational freedom | Head-group-specific or leaflet-specific chemistry |
| Which lipid species are changing? | Targeted or untargeted lipidomics | Class and species remodeling | Mobility, confinement, or direct mechanical state |
Comparison Table: Membrane Probes
| Probe | Main Readout | Strength | Limitation | Best Use Case |
|---|---|---|---|---|
| Laurdan | Interfacial hydration and dipolar relaxation | Strong for mapping membrane-state differences linked to hydration and phase behavior | Can be misread if hydration and polarity are not conceptually separated | Distinguishing membrane-state changes at the interface |
| DPH | Core order and rotational freedom | Established readout for hydrophobic-core ordering | Less informative for head-group or hydration effects | Tracking changes in acyl-chain order |
| DiI | Membrane visualization and partitioning tendency | Bright and practical for imaging-based membrane studies | Not a universal order metric | Visualizing membrane distribution and phase preference |
Composition and state must be read together
Membrane-state assays are strongest when they are not left alone. If FRAP suggests confinement, composition data help explain why. If Laurdan indicates a shift in hydration-sensitive order, lipidomics can reveal whether sterol redistribution, chain saturation, or phospholipid-class remodeling is a plausible driver. If a GUV experiment reveals phase separation, targeted compositional profiling can test whether the same lipid relationships appear in the source research model or membrane preparation.
This is where multi-layer assay design becomes more powerful than any single readout. A broad discovery lipid survey can map the lipid field, a targeted species-level lipid workflow can test the highest-value candidates, and a pathway-level interpretation of lipid remodeling layer can connect abundance shifts to membrane-state behavior rather than leaving them as disconnected observations.
Conclusion
Phospholipids should not be reduced to a list of membrane components. They are structural rules embedded in chemistry. Head-group area changes interfacial geometry. Acyl chains change packing, thickness, and transition behavior. Leaflet distribution changes which side of the membrane presents charge and which proteins can bind productively. Cholesterol and compatible lipids create ordered domains only under the right compositional conditions. Curvature-active lipids lower the energetic cost of topological change. Protein recruitment emerges from the combined logic of electrostatics, insertion energetics, ion shielding, and local membrane state.
Once this framework is in place, the membrane looks different. It is no longer a passive bilayer populated by proteins. It is an active physical platform whose composition determines what shapes, recruitment events, and compartment identities are possible. That is the architectural role of phospholipids.
The most useful membrane studies therefore do not ask for one universal assay. They match the measurement to the membrane question. Composition needs one kind of readout. Mobility needs another. Curvature logic may need a simplified model. Signaling-active phospholipid pools need deeper targeting than bulk totals can provide. In membrane research, the most reliable interpretation comes from reading phospholipid composition, membrane state, and assay limits together rather than treating any one readout as self-sufficient.
FAQ
Why does phosphatidylethanolamine promote curvature more strongly than phosphatidylcholine?
Because PE usually has a smaller effective head-group area relative to its hydrophobic region. That geometry makes it more cone-like and more compatible with negative curvature and high-curvature intermediates.
Why is phospholipid asymmetry considered an active property?
Because many phospholipids do not spontaneously cross the bilayer quickly. Cells use flippases, floppases, and scramblases to establish, maintain, or collapse leaflet differences as needed.
Why can small phosphoinositide pools have large organizational effects?
Because their function depends on local concentration and recognition specificity rather than bulk abundance. A small phosphoinositide-rich patch can recruit proteins and redefine membrane identity very efficiently.
Does cholesterol always make membranes more rigid?
No. Cholesterol acts in a composition-dependent way rather than as a one-way rigidifier. In more disordered, unsaturated membranes, it often condenses chain motion and promotes a more ordered liquid-like state. In already tightly packed saturated systems, it can instead disrupt overly regular packing and broaden otherwise sharp transitions. The outcome therefore depends on which lipid background cholesterol enters, not on cholesterol alone.
Why is lateral diffusion much faster than flip-flop?
Because lateral diffusion keeps the polar head group at the membrane interface, where it remains compatible with water. Flip-flop is much slower because the same head group must cross the hydrophobic core of the bilayer, which is energetically unfavorable. That barrier is one of the main reasons leaflet asymmetry can persist long enough to become biologically meaningful.
Why are GUVs useful if they are simpler than living cells?
Because they isolate membrane variables. That makes them ideal for testing phase behavior, curvature sorting, and probe response without the complexity of full cellular regulation.
Why can't Laurdan, DPH, and DiI be treated as interchangeable membrane probes?
Because they report through different physical mechanisms. Laurdan is strongly tied to interfacial hydration, DPH reports core order, and DiI-like dyes are often better for visualization than for universal order quantification.
Why can class totals miss important membrane remodeling?
Because large membrane-state shifts do not always require dramatic changes in class abundance. A membrane can remodel through changes in chain length, degree of unsaturation, molecular-species balance, or localized phosphoinositide pools while overall class totals appear relatively stable. This is why class-level summaries are often useful for orientation but insufficient for mechanism.
References
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112-124. DOI: 10.1038/nrm2330.
- Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 2018;19(5):281-296. DOI: 10.1038/nrm.2017.138.
- Levental I. Lipid rafts come of age. Nat Rev Mol Cell Biol. 2020. DOI: 10.1038/s41580-020-0252-x.
- Posor Y, Jang W, Haucke V. Phosphoinositides as membrane organizers. Nat Rev Mol Cell Biol. 2022;23(12):797-816. DOI: 10.1038/s41580-022-00490-x.
- Levental I, Lyman E. Regulation of membrane protein structure and function by their lipid nano-environment. Nat Rev Mol Cell Biol. 2023;24(2):107-122. DOI: 10.1038/s41580-022-00524-4.
- Nagata S, Sakuragi T, Segawa K. Regulation of phospholipid distribution in the lipid bilayer by flippases and scramblases. Nat Rev Mol Cell Biol. 2023. DOI: 10.1038/s41580-023-00604-z.
- Nair KS, Bajaj H. Advances in giant unilamellar vesicle preparation techniques and applications. Adv Colloid Interface Sci. 2023;318:102935. DOI: 10.1016/j.cis.2023.102935.
*Note: This resource discusses phospholipid behavior, membrane-state readouts, and analytical workflows in research-use-only (RUO) membrane models and experimental systems, not in clinical, diagnostic, or therapeutic contexts.
