What Is Venom Peptide Peptidomics?
Venomous animals — including cone snails, spiders, scorpions, and snakes — produce complex peptide cocktails that have evolved over millions of years to selectively target specific receptors, ion channels, and enzymes in prey and predators. These venom peptides represent one of nature's most concentrated libraries of bioactive scaffolds, many of which show exceptional affinity and selectivity for drug targets that are difficult to address with small molecules or large biologics.
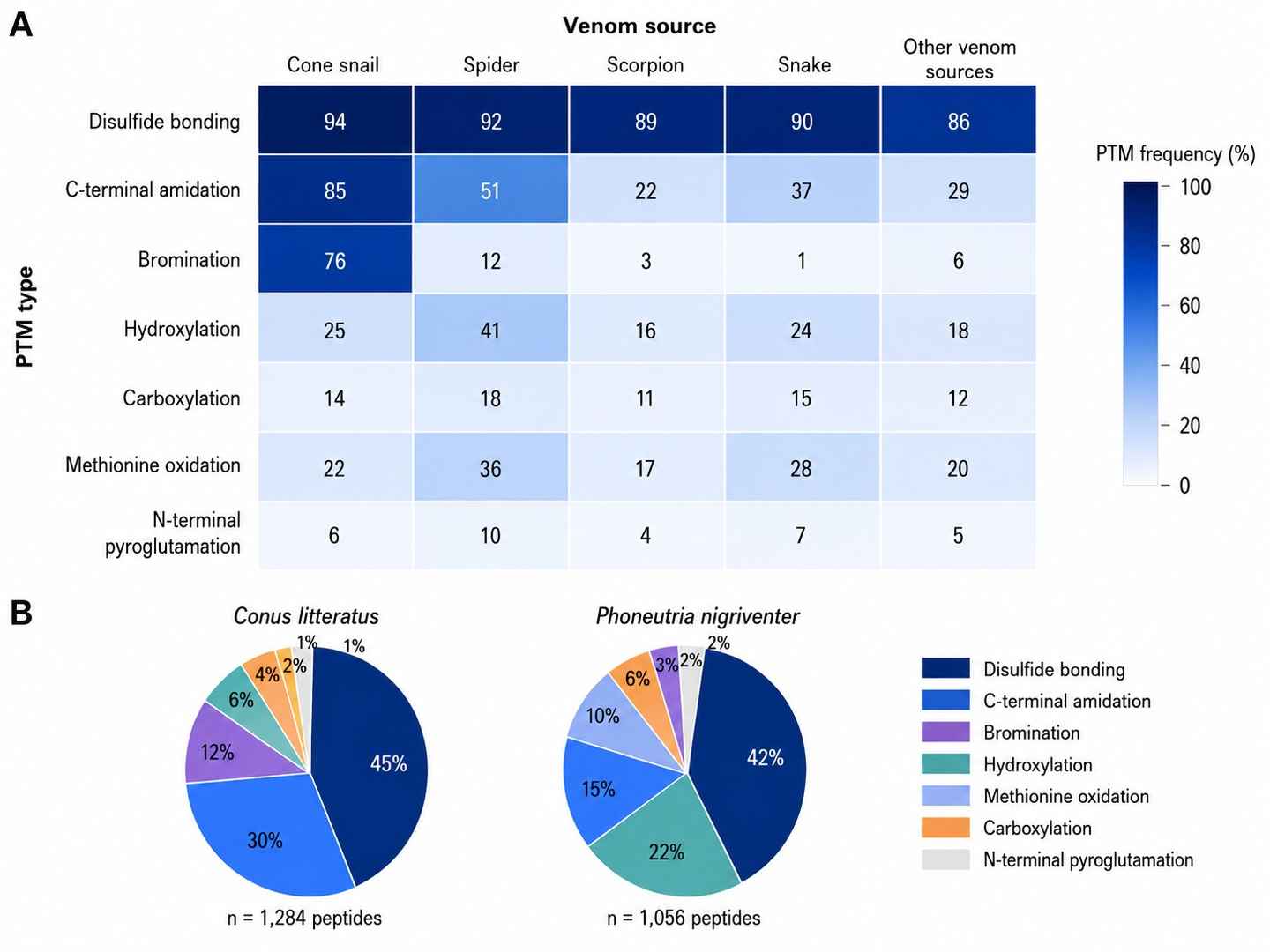
Venom peptide peptidomics applies LC-MS/MS-based mass spectrometry to systematically identify, characterize, and quantify the peptide components of animal venoms. Unlike standard proteomics workflows, venom peptidomics must contend with extreme peptide density, a high proportion of cysteine-rich sequences stabilized by multiple disulfide bonds, and diverse post-translational modifications — including bromination, hydroxylation, and carboxylation — that are critical to bioactivity.
The field bridges two powerful research traditions: the pharmacological exploration of venom as a source of therapeutic leads, and the analytical rigor of modern mass spectrometry-based peptidomics. For researchers working at this intersection, a specialized venom peptide analysis platform is essential for capturing the full diversity of venom components and translating that diversity into actionable drug discovery data.
Venom Peptide Characterization Services — What We Offer
The Creative Proteomics venom peptide peptidomics platform is purpose-built for the analytical challenges that venom matrices present. Nine integrated service modules cover the complete journey from crude venom input to publication-ready peptide identification data, without requiring researchers to piece together multiple service providers.
Learn more about our specialized venom peptide services: GPCR-targeted venom peptide screening, ion channel profiling, cone snail conotoxin characterization, and venom peptide lead optimization.
Detectable Venom Peptide Types and Species Coverage
The platform accommodates venom peptide analysis across all major venomous animal classes and their characteristic peptide families.
| Venom Source | Representative Peptide / Toxin Families | Key Structural Features |
|---|---|---|
| Cone snails (Conus spp.) | Conotoxins (T1, T2, A, M, O1, P, I1, I2, I3, J superfamilies), conantokins, conopressins | Multiple disulfide frameworks (IC, IIC, III, IV, VI/VII); bromination, C-terminal amidation |
| Spiders (Myrmekiagen, Phoneutria, Atrax) | Cysteine-rich knot toxins, linear pore-forming peptides, CRISP proteins | Inhibitor cystine knot (ICK) motif; disulfide-stabilized three-dimensional scaffolds |
| Scorpions (Buthus, Tityus, Androctonus) | Potassium channel toxins (α-KTx), sodium channel toxins (β-NaKTx), antimicrobial peptides | α-helical disulfide-stabilized folds; charybdotoxin and maurotoxin frameworks |
| Snakes (Bothrops, Crotalus, Naja) | Three-finger toxins, PLA2 enzymes, metalloproteases, serine proteases | Three-finger fold; disulfide-dense scaffolds; high target specificity |
| Other venom sources | Centipedes, wasps, ants, jellyfish, sea anemones | Diverse scaffolds including linear amphipathic peptides and disulfide-rich toxins |
Notes:
- The platform supports peptide mass range of 500–8,000 Da, optimized for the venom peptide size distribution.
- PTM detection includes disulfide mapping, oxidation, bromination, hydroxylation, carboxylation, and C-terminal amidation.
- For venom species not listed here, contact us to discuss feasibility prior to sample submission.
Unified Venom Peptide Peptidomics Workflow
The Creative Proteomics venom peptide peptidomics platform integrates six analytical stages — from venom extraction through data reporting — into a single cohesive project structure.
Venom Peptide Extraction
RP-HPLC fractionation and partial reduction-alkylation
LC-MS/MS Analysis
Orbitrap or timsTOF — DDA or DIA acquisition
PTM Mapping and Database Search
UniProt toxin + venom gland transcriptome hybrid search
Disulfide Connectivity Mapping
Partial reduction MS/MS — up to four disulfide bonds
Quantitative Venom Peptidomics
LFQ, TMT/iTRAQ, or PRM/MRM quantification
Data Reporting
Peptide inventory, PTM maps, quantitative tables, MS/MS spectra
1
Venom Peptide Extraction
Venom samples are fractionated using reversed-phase HPLC to separate peptides from proteins and small molecules. For disulfide-rich toxins, a partial reduction-alkylation step is applied prior to separation to preserve information about native disulfide connectivity while enabling successful MS/MS fragmentation of cysteine-dense sequences.
2
LC-MS/MS Analysis
Fractionated peptides are analyzed on a high-resolution Orbitrap or timsTOF instrument operating in data-dependent acquisition (DDA) or data-independent acquisition (DIA) mode. MS1 spectra are collected at high resolving power (≥60,000 at m/z 200) to accurately determine peptide mass, while MS/MS spectra provide fragment ion information for de novo sequencing and database-matched identification.
3
PTM Mapping and Database Search
Tandem mass spectra are searched against a hybrid database combining known venom protein sequences (UniProt and specialized toxin databases) and theoretically predicted peptide sequences from venom gland transcriptomes. Searches are configured for variable modifications including disulfide formation (Cys-Cys), methionine oxidation, proline hydroxylation, tryptophan bromination, and glutamate carboxylation.
4
Disulfide Connectivity Mapping
For cysteine-rich venom peptides — the majority of pharmacologically active conotoxins, spider toxins, and scorpion venom components — Creative Proteomics applies a specialized disulfide mapping workflow. Partial reduction under controlled conditions generates subsets of peptide species with known numbers of intact disulfide bonds, and MS/MS analysis of each subset determines which cysteine residues are paired. This approach resolves connectivity in peptides with up to four disulfide bonds without requiring NMR or X-ray crystallography.
5
Quantitative Venom Peptidomics
Relative quantification across venom samples, individual variation, or treatment conditions is performed using label-free quantification (LFQ) or isobaric labeling (TMT or iTRAQ). For absolute quantification of specific venom peptides, a parallel reaction monitoring (PRM) or multiple reaction monitoring (MRM) assay is developed on a triple quadrupole or Orbitrap platform.
6
Data Reporting
Peptide identification results are compiled with FASTA sequence files, MS/MS spectra for key identifications, PTM localization confidence scores, quantitative comparison tables, and a searchable venom peptide inventory. All data are delivered in formats suitable for downstream functional screening, structural biology, or manuscript preparation.
Deep and Accurate Venom Peptide Detection
Creative Proteomics addresses the specific analytical challenges that venom matrices present — high disulfide density, PTM diversity, and extreme peptide concentration range — with purpose-developed sample preparation protocols and instrument configurations tuned for the venom peptide mass range.
Why Venom Peptidomics Requires Specialized Workflows
Standard bottom-up proteomics workflows — optimized for cell lysates or tissue samples — frequently underperform on venom matrices for three structural reasons:
- High disulfide density: Venom peptides often contain 2–6 disulfide bonds per molecule, which constrain peptide fragmentation in MS/MS if not selectively reduced. Generic reduction-alkylation destroys connectivity information entirely.
- PTM diversity: Non-ribosomal PTMs such as bromination (critical for bioactivity in many conotoxins), hydroxylation, C-terminal amidation, and sulfation are not included in standard search databases. Targeted modification databases are required.
- Extreme peptide concentration range: A single venom can contain major peptide components at µg/mL levels alongside minor components at ng/mL or lower. Detecting both requires a dynamic range that pushes the limits of most LC-MS/MS platforms.
| Parameter | Specification |
|---|---|
| Mass range | 500–8,000 Da (optimized for venom peptides) |
| MS1 resolution | ≥60,000 at m/z 200 (Orbitrap) |
| MS/MS fragmentation | HCD and CID for peptide sequencing |
| Acquisition modes | DDA and DIA available |
| Sample input | ≥1 µg total venom for comprehensive profiling |
| PTM detection | Disulfide mapping, oxidation, bromination, hydroxylation, carboxylation |
| Quantification | Label-free (LFQ), TMT/iTRAQ, PRM/MRM |
| Database | UniProt toxin sequences + venom gland transcriptomes |
Why Choose Creative Proteomics for Venom Peptide Analysis
Sample Requirements for Venom Peptide Peptidomics
| Parameter | Specification |
|---|---|
| Sample type | Crude venom (milkable or dissected venom glands) |
| Sample form | Lyophilized powder or fresh-frozen in buffer-free tube |
| Minimum input | 1 µg total venom (standard profiling); 5–10 µg (disulfide mapping) |
| Purity | Crude venom acceptable; do not pre-fractionate or remove proteins |
| Storage | Ship on dry ice; store at −80°C upon receipt |
| Shipping | FedEx/UPS overnight with dry ice tracking |
| Volume | 10–50 µL crude venom or equivalent lyophilized mass |
| Biological replicates | Recommend ≥3 per condition for comparative studies |
| Metadata required | Species name (scientific), geographic origin, specimen condition, extraction date |
| Safety documentation | Material Safety Data Sheet (MSDS) for all venom samples required at booking |
Note: Venom samples from protected or regulated species require appropriate collection permits. Please confirm sourcing compliance before submitting samples.
Deliverables | What You Will Receive
- Comprehensive Venom Peptide Inventory
FASTA format with peptide sequences, masses, and PTM annotations for all identified venom components. - PTM Localization Report
Confidence scores for bromination, hydroxylation, carboxylation, C-terminal amidation, and other venom-specific modifications. - Disulfide Connectivity Map
Cysteine pairing assignments for cysteine-rich venom peptides — up to four disulfide bonds resolved via partial reduction MS/MS. - MS/MS Spectra Archive
Raw fragment ion spectra for key peptide identifications, delivered in open formats for independent review. - Quantitative Comparison Tables
LFQ, TMT/iTRAQ, or PRM/MRM quantification results across samples, conditions, or species. - Search Parameters and FDR Statistics
Complete database search configuration, false discovery rate estimates, and QC metrics. - Bioinformatics Support
Pathway annotation, toxin family classification, and cross-species comparative analysis. - Publication-Ready Figures and Data Tables
Formatted for direct inclusion in manuscripts and grant applications.
Representative Data
Venom Peptide Identification Coverage — Conus litteratus
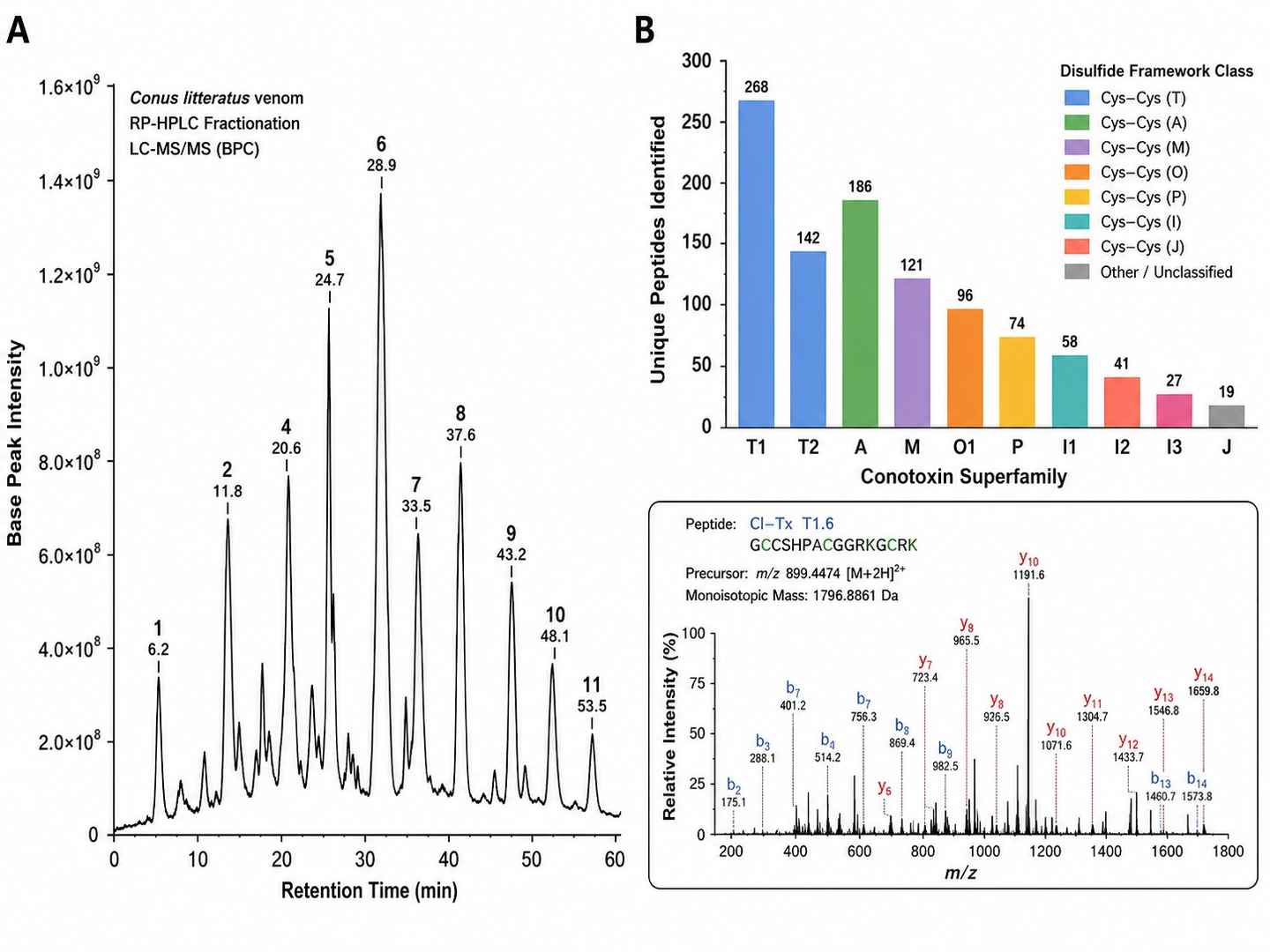
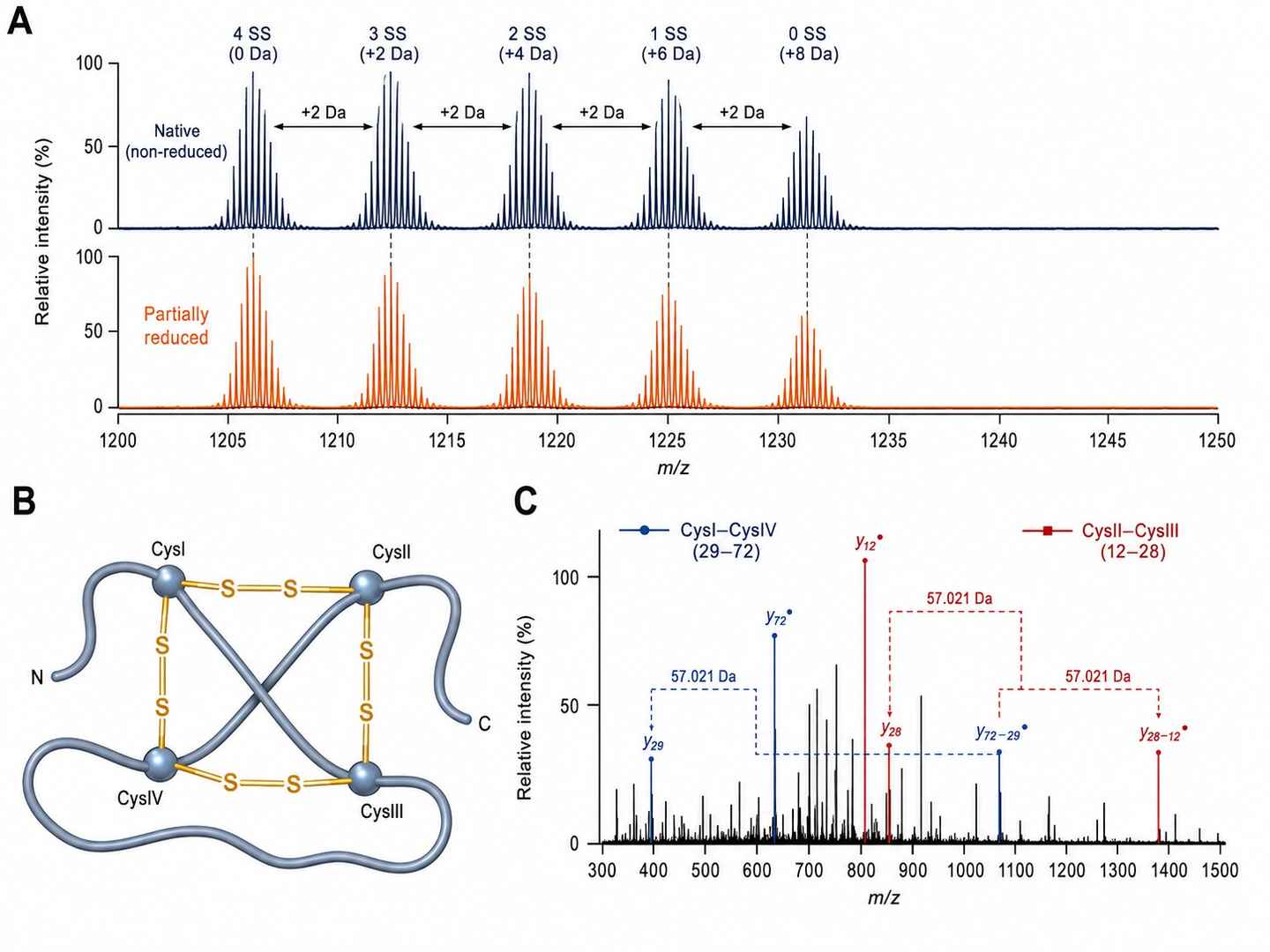
Disulfide Connectivity Mapping — ICK Framework Toxin
PTM Distribution Across Venom Peptide Classes
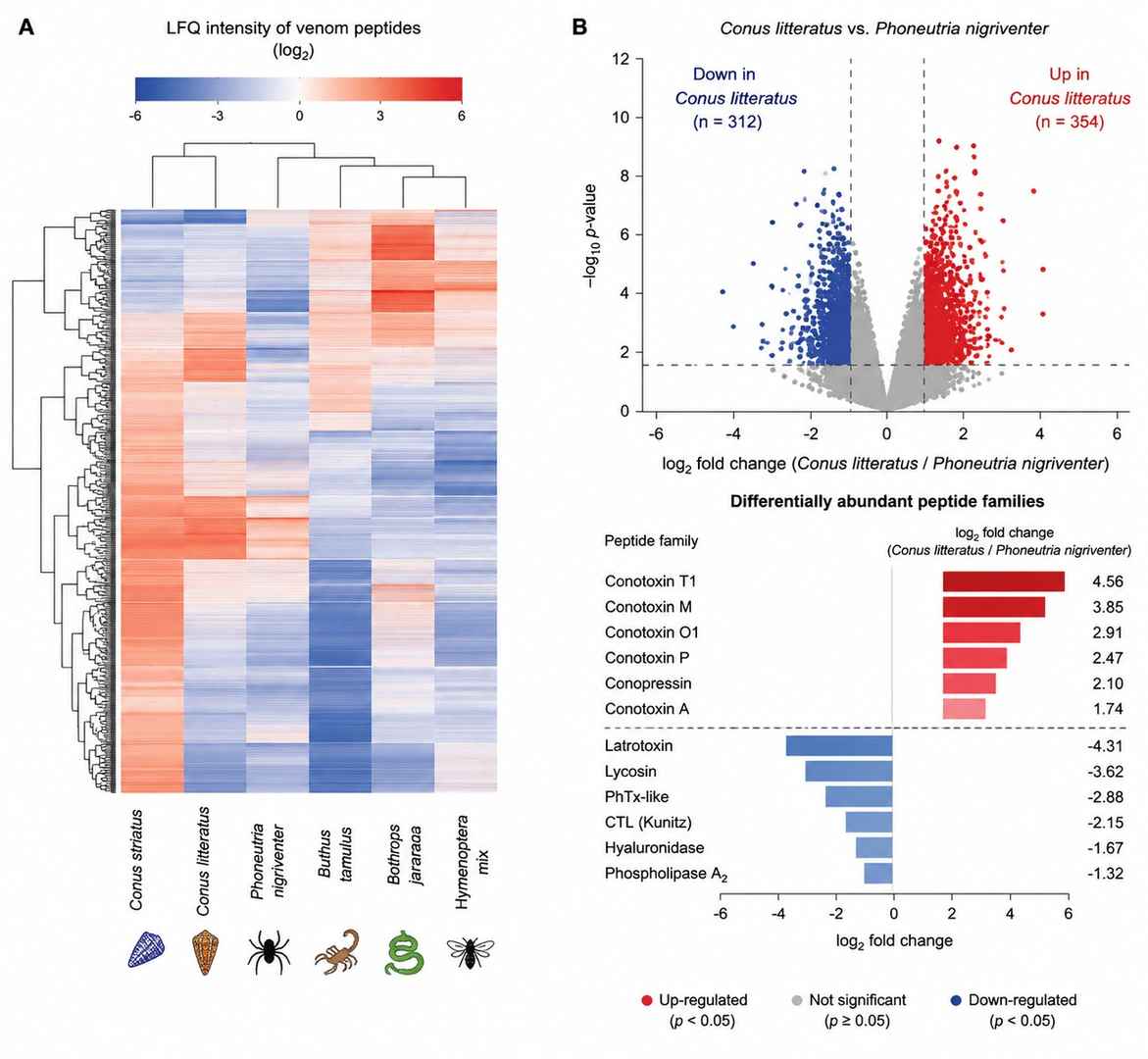
Comparative Venom Peptidomics — Interspecies LFQ
What venom species can be analyzed by the Venom Peptide Peptidomics Service? +
The platform supports venom peptide analysis across all major venomous animal classes: cone snails (Conus spp.), spiders (Myrmekiagen, Phoneutria, and related genera), scorpions (Buthus, Tityus, Androctonus), snakes (Bothrops, Crotalus, Naja, and related genera), and additional venom sources including centipedes, wasps, ants, jellyfish, and sea anemones. For venom species not listed here, contact us to discuss feasibility prior to sample submission.
What is the minimum venom sample input required? +
The standard service requires a minimum of 1 µg of total venom material for comprehensive peptide profiling. For disulfide connectivity mapping or targeted quantification of specific low-abundance venom peptides, we recommend providing 5–10 µg to ensure sufficient coverage. If your available sample is below these thresholds, contact us — specialized low-input protocols are available for certain venom types.
How does the service handle disulfide-rich venom peptides? +
Disulfide-rich venom peptides are addressed through a partial reduction-alkylation workflow prior to LC-MS/MS analysis. Under controlled reduction conditions, a subset of disulfide bonds remains intact while others are selectively reduced and alkylated. Comparing MS/MS data from reduced-alkylated versus native (non-reduced) samples allows us to determine the number of intact disulfide bonds and — through iterative analysis of partially reduced species — assign specific cysteine pairings. This approach supports peptides with up to four disulfide bonds.
What PTMs are detected in the venom peptide analysis? +
The standard venom peptide PTM panel includes: methionine oxidation, proline hydroxylation, tryptophan bromination, glutamate carboxylation, C-terminal amidation, N-terminal pyroglutamation, and disulfide bond formation (Cys-Cys). For specific venom sources known to carry unusual modifications (e.g., sulfation, glycosylation, deamidation), additional variable modifications can be configured in the database search. If you are looking for a specific PTM not listed here, please specify it in your inquiry.
Can the service support comparative venom profiling across multiple samples? +
Yes. Quantitative comparative venomomics is available using label-free quantification (LFQ) or isobaric labeling (TMT) for relative comparison, and PRM/MRM for targeted absolute quantification of specific venom peptides. Comparative studies can span geographic populations of the same species, individual venom variation, or changes in venom composition in response to environmental or physiological conditions.
Is the venom peptide identification data suitable for publication? +
Yes. All deliverables are provided in formats consistent with journal requirements for proteomics data (IMProV, MCP, JPR). Peptide identification results include FASTA files, search parameters, FDR estimates, PTM localization scores, and MS/MS spectra for key peptide identifications. Our QC pipeline applies a 1% FDR threshold at the peptide level.
Case Study — AI-Guided Venom Peptide Library Design Meets MS Characterization
Journal: Pharmaceuticals (MDPI)
Published: 2026
Summary
Traditional venom peptide drug discovery has been constrained by limited natural sequence diversity — researchers have relied on whatever peptides exist in the venom of collected specimens, with no systematic way to explore the mutational space around a given bioactive scaffold. This limitation is particularly acute for challenging drug targets like the Notch ligand DLL3, where conventional phage display libraries fail to yield high-affinity binders. Cai et al. (2026) addressed this gap by combining a large-scale, venom-derived peptide library (VCX) with machine learning-guided scaffold optimization and high-throughput MS-validated expression.
Methods
The team constructed a venom peptide display (VCX) library containing approximately 482 distinct conotoxin and spider toxin scaffolds, generating approximately 6 × 10⁵ unique peptide members. Scaffold selection was guided by a RoBERTa-based transformer model trained to predict peptide fold stability — allowing researchers to computationally identify residue positions that tolerate mutation without compromising the disulfide-stabilized core structure. Phage display p3 and p8 systems were used for biopanning against four target proteins: CD47, DLL3, IL33, and P2X7R. Hit validation employed surface plasmon resonance (SPR) for affinity measurement and LC-MS to confirm expression integrity of Trx-fusion venom peptide constructs. Affinity maturation used a single-cycle NNK-block saturation mutagenesis strategy guided by ML fold prediction.
Key Findings
| Metric | Value |
|---|---|
| Library size | ~6 × 10⁵ unique peptides |
| Scaffold diversity | 482 distinct venom peptide scaffolds |
| VCX fold score (median) | 0.8606 vs. 0.7028 for control library |
| Screening targets | CD47, DLL3, IL33, P2X7R |
| Hit rate (targets with binders) | 4/4 (100%) |
| DLL3 affinity — initial hit | ~3.5 µM |
| DLL3 affinity — after ML-guided maturation | 6.7 nM and 12.6 nM |
| Affinity improvement | ~500-fold in single maturation cycle |
| Recombinant expression yield (Trx-VCX constructs) | 77% >20 mg/L; 16% >100 mg/L |
What This Means for Your Venom Peptide Drug Discovery Program
- A structurally diverse venom peptide library (482 scaffolds) provides a broader starting point than conventional random or consensus libraries for targeting difficult proteins including GPCRs and ion channels
- Machine learning-guided fold prediction enables rational exploration of mutational space around disulfide-rich venom scaffolds, predicting which positions tolerate substitution without disrupting the bioactive conformation
- A single-cycle ML-assisted affinity maturation achieved approximately 500-fold affinity improvement for DLL3 — comparable to results that typically require 2–3 rounds of traditional directed evolution
- High-throughput recombinant expression of venom peptide Trx-fusion constructs in E. coli demonstrates that venom scaffold diversity is compatible with standard microbial expression systems, supporting rapid lead production
- The integrated phage display + MS validation + SPR characterization pipeline bridges discovery, expression, and functional validation in a continuous workflow
- The 100% screening hit rate across four mechanistically diverse targets (immune checkpoint, cytokine, ion channel, membrane receptor) suggests broad applicability of the VCX platform across therapeutic areas
Conclusion
The convergence of large-scale venom peptide library design, machine learning-assisted scaffold optimization, and MS-validated expression represents a step-change in the efficiency of venom-based peptide drug discovery. By systematically expanding the mutational space around naturally evolved bioactive scaffolds — and validating results through orthogonal biophysical methods — researchers can move from initial venom screening to high-affinity, developable lead candidates in a fraction of the time required by conventional approaches.
References
- Cai F, Zhou L, Delgado B, et al. A Machine Learning-Enabled Venom Peptide Platform for Rapid Drug Discovery. Pharmaceuticals. 2026;19(2):288. https://doi.org/10.3390/ph19020288
- Abd El-Aziz TM, Soares AG, Stockand JD. Advances in venomics: Modern separation techniques and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1160:122352. https://doi.org/10.1016/j.jchromb.2020.122352
- Mouchbahani-Constance S, Sharif-Naeini R. Proteomic and Transcriptomic Techniques to Decipher the Molecular Evolution of Venoms. Toxins. 2021;13(2):154. https://doi.org/10.3390/toxins13020154
*All services are for research use only (RUO). Not for diagnostic or clinical use.*