What Is Endogenous Peptidomics?
Endogenous peptides are short, bioactive molecules generated by the proteolytic cleavage of precursor proteins within living systems. This diverse class includes neuropeptides, peptide hormones, antimicrobial peptides, cytokine fragments, and collagen-derived signaling molecules — each playing distinct roles in intercellular communication, metabolic regulation, and physiological homeostasis.
Unlike conventional proteomics, which relies on enzymatic digestion (typically trypsin) to generate analyzable peptides, endogenous peptidomics targets the naturally occurring peptide repertoire directly — without artificial proteolysis. This distinction is critical: enzymatic digestion destroys the very molecular signals that living systems use for signaling and regulation, making it impossible to study the true bioactive peptide landscape through standard proteomic workflows.
Creative Proteomics has built a dedicated endogenous peptidomics platform to address this analytical gap. From low-volume biofluid samples to multi-milligram tissue specimens, the platform captures naturally processed peptides across the full mass range, resolves post-translational modifications (PTMs) at single-amino-acid resolution, and delivers quantitative readouts suitable for biomarker discovery, mechanism research, and drug target identification.
Endogenous Peptide Profiling Services — What We Offer
Creative Proteomics delivers a comprehensive endogenous peptide profiling service, combining optimized peptide enrichment, high-resolution LC-MS/MS acquisition, and PTM-aware bioinformatics to characterize the complete bioactive peptide landscape of any biological sample.
Detectable Endogenous Peptide Types and Sample Coverage via LC-MS/MS
| Endogenous Peptide Category | Representative Peptides | Representative Biofluid / Tissue Sources |
|---|---|---|
| Neuropeptides | Substance P, β-endorphin, NPY, Dynorphin A, Somatostatin, Oxytocin, CRF, CART | Brain tissue, CSF, plasma — see also Neuropeptidomics Service |
| Peptide Hormones | GLP-1, Insulin, Glucagon, Leptin, Ghrelin, Adiponectin fragments, GIP | Plasma, serum, pancreatic tissue |
| Cardiovascular Peptides | ANP, BNP, Endothelin-1, Adrenomedullin, Apelin | Plasma, cardiac tissue, vascular endothelium |
| Gastrointestinal Peptides | CCK-8, Gastrin, Secretin, VIP, PACAP, Motilin | GI tissue, gastric juice, plasma |
| Antimicrobial Peptides (AMPs) | LL-37, α-Defensins, β-Defensins, Cathelicidin, Lactoferrin fragments | Saliva, tears, skin, plasma, urine — see also AMP and HDP Profiling Service |
| Collagen-Derived Peptides | Prolyl-hydroxyproline (Hyp-Gly), Type I/III collagen fragments, N-acetylated peptides | Plasma, serum, bone, cartilage, skin tissue |
| Muscle-Wasting and Myokine Peptides | Myostatin fragments, Irisin, Meteorin-like peptide fragments | Skeletal muscle tissue, plasma |
| Enzyme-Cleavage Products | Fibrinopeptide A/B, Fibrin degradation products, D-dimer fragments | Plasma, serum |
| Cytokine and Chemokine Fragments | IL-1β fragments, CXCL8 truncations, TGF-β latent complex peptides | Plasma, tissue culture supernatant, inflammatory fluids |
| Cryptic Peptides (De Novo Discovery) | Peptides from non-canonical cleavage, alternative reading frames, or untranslated regions | Any biofluid or tissue — see also RiPP Genome Mining Service |
Notes:
- Detection supports modified forms: C-terminal amidation, N-terminal pyroglutamation, disulfide bonds, hydroxylation, bromination, oxidation, and phosphorylation.
- Peptides can be mapped back to precursor or prohormone proteins (e.g., POMC, NPY, proinsulin, proglucagon, cathelicidin precursor).
- Coverage includes both canonical bioactive peptides and cryptic or species-specific peptides identified through open-search and de novo sequencing pipelines.
Endogenous Peptidomics Platform Technology Highlights
- High-Resolution LC-MS/MS Platform
High-resolution nanoLC coupled to Q-TOF or Orbitrap mass spectrometry systems (Q Exactive HF-X, Orbitrap Exploris™ 480, Orbitrap Astral™, timsTOF Pro) for deep peptide coverage across the endogenous mass range. - PTM-Aware Search Engine with De Novo Assist
Combines database search with open-modification workflows and de novo sequencing to identify both catalogued and previously uncharacterized endogenous peptides, including those with atypical cleavage sites or non-canonical modifications. - Molecular-Weight Cutoff + C18 SPE Dual Enrichment
Endogenous peptides are enriched by 30 kDa MWCO filtration to remove high-MW proteins, followed by C18 solid-phase extraction (SPE) to desalt and concentrate low-MW peptide fractions. Dual-enrichment minimizes high-abundance protein contamination and maximizes recovery of short, bioactive peptides. - Acidic and Protease-Inhibiting Extraction Conditions
All sample preparation is performed under acidic conditions (typically 0.1–1% TFA or formic acid) with protease inhibitor cocktails to halt endogenous proteolytic activity and preserve the authentic in-vivo peptide profile at the moment of collection. - Label-Free Quantitation with QC Normalization and Batch Correction
MS1 peak-area–based label-free quantitation is normalized using pooled QC samples injected at regular intervals throughout the acquisition batch. EigenMS or similar batch-effect correction algorithms are applied to remove systematic drift, enabling cross-batch comparability for multi-cohort studies. - Bioinformatics with Functional Annotation and Protease Inference
Peptide sequences are annotated for source proteins, GO terms, and KEGG pathways. Cleavage site patterns (p4–p4' window analysis) are computed to infer the active protease repertoire contributing to the observed peptide landscape.

Endogenous Peptidomics Platform Specifications
| Feature / Specification | Creative Proteomics Platform |
|---|---|
| Analysis platform | High-resolution LC-MS/MS (Q-TOF, Orbitrap, timsTOF) |
| Search engine | PTM-aware engine + de novo sequencing assist |
| PTM coverage | C-terminal amidation, N-terminal pyroglutamation, disulfide connectivity, hydroxylation, bromination, oxidation, phosphorylation |
| Plasma single-run ID depth | 5,000–20,000+ peptide IDs per sample (biofluid) |
| Minimum sample input | Plasma 50 µL; urine 10 mL; CSF 150 µL; tissue 50 mg |
| Quantitative method | Label-free MS1 peak area + QC internal standard normalization + EigenMS batch correction |
| Bioinformatics deliverables | Peptide inventory, PTM annotation table, quantitative tables, PCA / volcano plots, GO/KEGG enrichment, protease cleavage site profiling |
| Deliverable format | RAW data + PDF report + raw data tables (Excel) |
| Sample type coverage | Plasma, serum, urine, CSF, saliva, tissue homogenate, cell secretome — 10+ types |
Why Choose Creative Proteomics for Endogenous Peptidomics LC-MS/MS Analysis
Unified Endogenous Peptidomics Workflow
Sample Receipt and Quality Check
Sample integrity verification, peptide / protein content estimation, and sample-type-specific storage confirmation.
Peptide Extraction and Enrichment
30 kDa MWCO filtration and C18 SPE under acidic conditions with protease inhibitors to enrich endogenous peptides and remove interfering high-MW proteins.
LC-MS/MS Data Acquisition
High-resolution nanoLC-MS/MS using Q-TOF or Orbitrap systems; DDA, DIA, or targeted acquisition modes selected based on project goals.
Database Search and PTM Annotation
PTM-aware database search combined with de novo sequencing for known and novel endogenous peptide identification and PTM site localization.
Label-Free Quantitation and QC Normalization
MS1 peak-area extraction, QC internal standard normalization, EigenMS batch-effect correction, and statistical analysis across conditions.
Report Generation
Comprehensive peptide inventory, PTM annotation, quantitative tables, functional enrichment, and PDF analysis report.
1
Sample Receipt and Quality Check
All submitted samples undergo initial integrity verification and peptide / protein content estimation. Storage conditions are confirmed to match sample-type requirements. This step ensures input material is suitable for low-MW peptide extraction and downstream LC-MS/MS analysis.
2
Peptide Extraction and Enrichment
Endogenous peptides are enriched by 30 kDa molecular-weight cutoff (MWCO) filtration to remove high-MW proteins, followed by C18 solid-phase extraction (SPE) for desalting and concentration. All steps are performed under acidic conditions (0.1–1% TFA or formic acid) with protease inhibitor cocktails to prevent post-collection proteolysis and preserve the authentic in-vivo peptide profile.
3
LC-MS/MS Data Acquisition
Enriched peptide fractions are separated by nanoLC and analyzed by high-resolution tandem mass spectrometry (Q-TOF, Orbitrap Exploris™ 480, Orbitrap Astral™, or timsTOF Pro). Acquisition modes (DDA, DIA, or targeted PRM) are selected to match project goals for depth, quantitative precision, or specific peptide targets.
4
Database Search and PTM Annotation
Peptide identification combines PTM-aware database search with de novo sequencing to capture both catalogued and previously uncharacterized endogenous peptides. Modifications including C-terminal amidation, disulfide connectivity, pyroglutamation, hydroxylation, bromination, oxidation, and phosphorylation are annotated and localized. Precursor protein mapping links each peptide back to its prohormone or source protein.
5
Label-Free Quantitation and QC Normalization
Peptide abundance is quantified using MS1 peak-area integration. Pooled QC samples injected at regular intervals enable intensity-based normalization, and EigenMS or equivalent batch-effect correction removes systematic drift between runs. Resulting quantitative data is suitable for PCA, volcano plot analysis, and clustering across biological conditions.
6
Report Generation
Final deliverables include the complete peptide identification list with PTM annotations, quantitative comparison tables, PCA / volcano / clustering visualizations, GO / KEGG functional enrichment, protease cleavage site analysis, and a comprehensive PDF report formatted for publication or regulatory submission.
Sample Requirements for Endogenous Peptidomics
| Sample Type | Minimum Amount | Recommended Amount | Preservation Method | Shipping Condition | Notes |
|---|---|---|---|---|---|
| Plasma (EDTA or heparin) | 50 µL | 200 µL | Aliquot in low-protein-bind tubes; snap-freeze | Dry ice | Avoid repeated freeze-thaw cycles; protease inhibitors recommended |
| Serum | 50 µL | 200 µL | Collect in serum-separating tubes; aliquot and freeze immediately | Dry ice | Clot formation must be complete before freezing |
| Urine | 10 mL | 50 mL | Mid-stream collection preferred; protease inhibitors added; aliquot | Dry ice | Morning first-void urine recommended for highest peptide yield |
| Cerebrospinal fluid (CSF) | 150 µL | 500 µL | Aliquot in low-protein-bind tubes; snap-freeze immediately | Dry ice | Use protease inhibitors; avoid repeated freeze-thaw |
| Saliva | 500 µL | 1 mL | Collect in saliva collection devices; protease inhibitors; centrifuge to remove particulates | Dry ice | Avoid food intake 30 min before collection |
| Tissue (brain, liver, kidney, lung) | 50 mg | 200 mg | Snap-freeze immediately after dissection; no fixatives or embedding | Dry ice | Store at –80°C immediately after dissection; record wet weight |
| Tissue (bone, cartilage, skin) | 500 mg | 1 g | Snap-freeze; decalcification required for bone before processing | Dry ice | Consult before submission for hard-tissue sample preparation guidance |
| Cell secretome / conditioned medium | 25 mL | 50 mL | Centrifuge to remove cells; filter through 0.22 µm; add protease inhibitors; freeze | Dry ice | Record cell count and confluence at collection; serum-free medium preferred |
| Bacterial / fungal biomass | 150 mg | 500 mg | Harvest mid-log phase; wash with PBS; snap-freeze | Dry ice | Record growth phase and culture conditions |
Representative Endogenous Peptidomics Data
Peptide Identification Coverage Across Biological Matrices
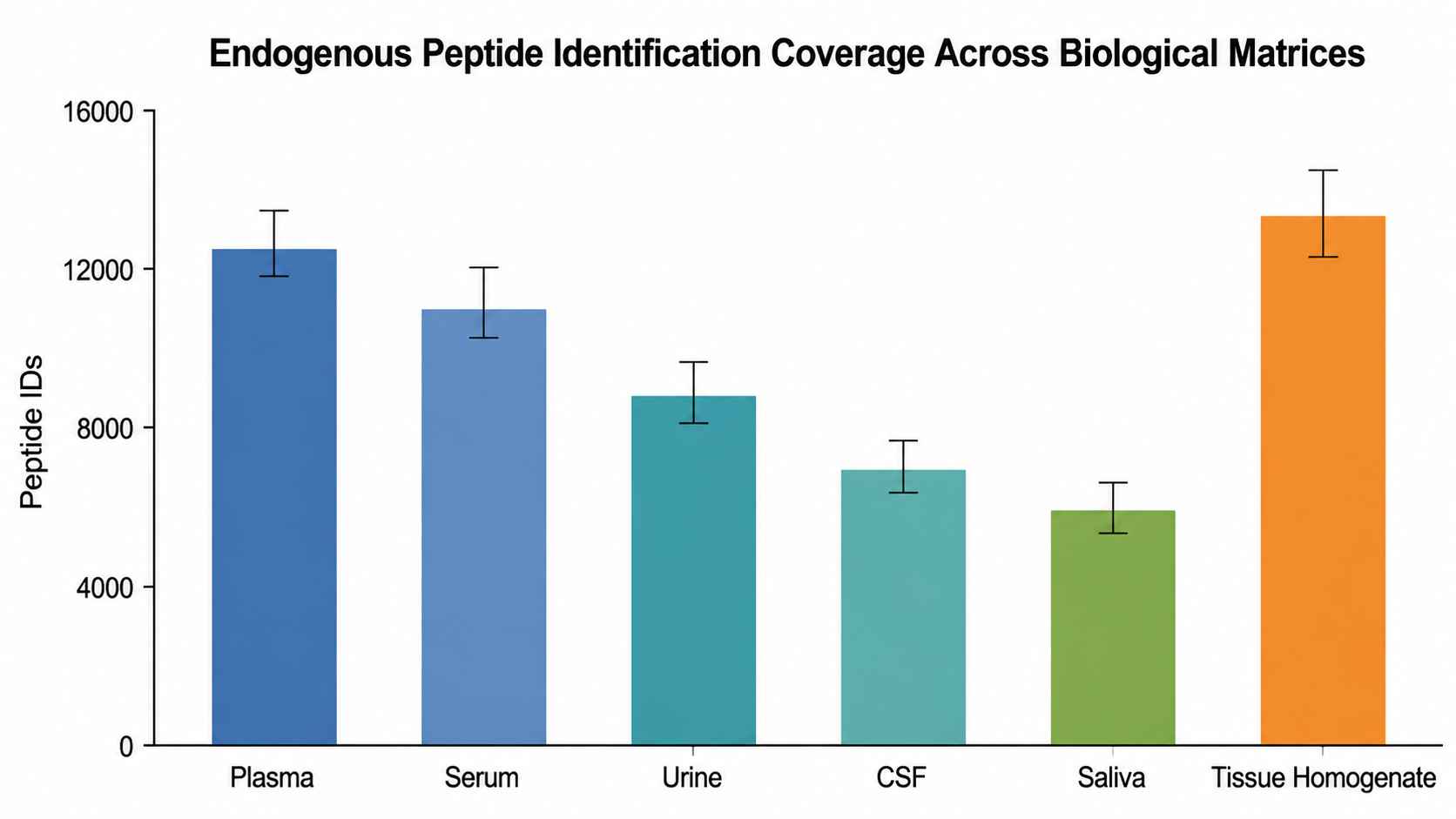
Endogenous peptide identification depth across diverse biological matrices. Bars represent total peptide IDs per sample type, demonstrating platform coverage from low-volume biofluids (50 µL plasma) to multi-milligram tissue specimens.
PTM Distribution Across Endogenous Peptide Classes
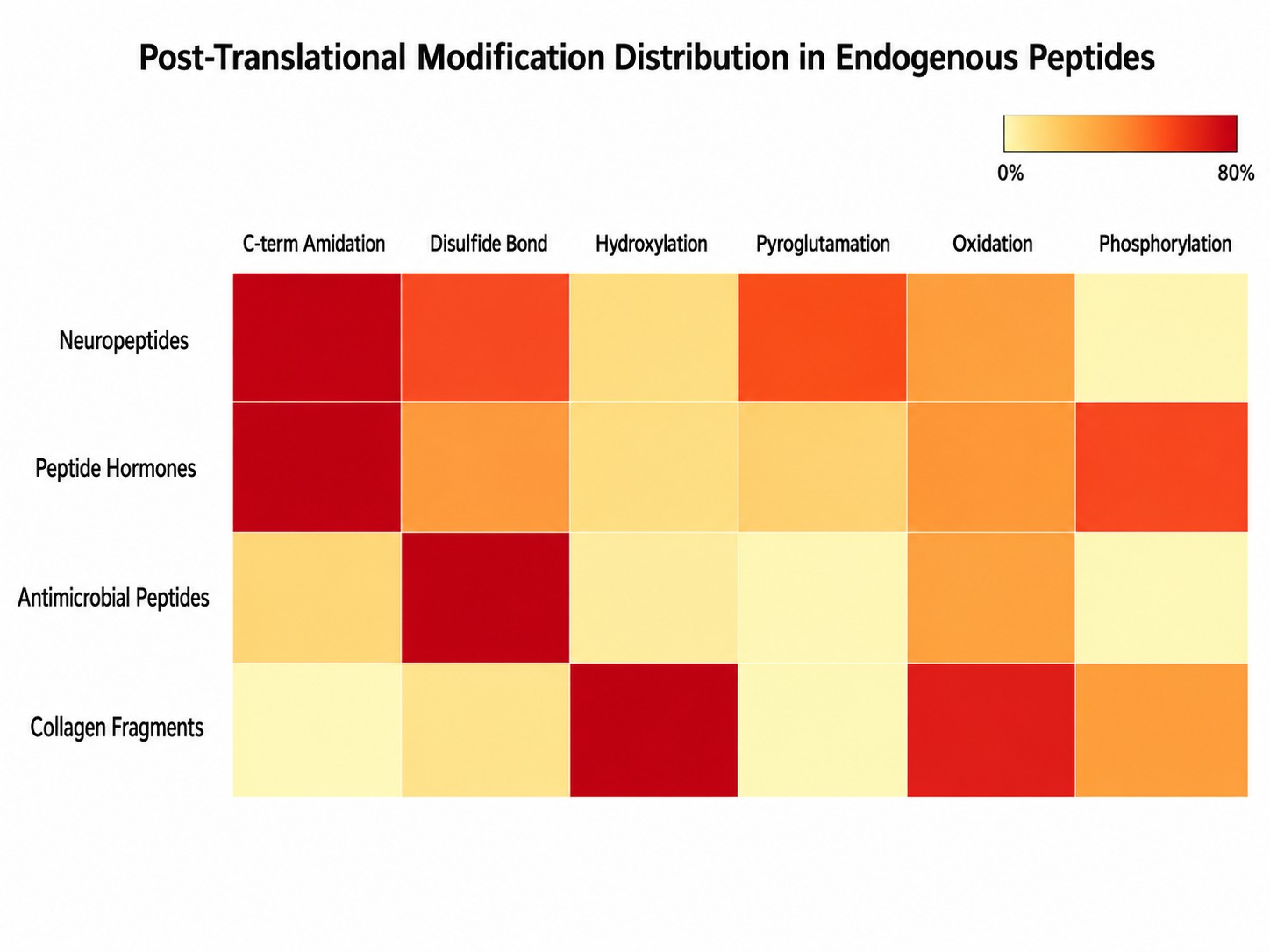
Post-translational modification prevalence across major peptide classes — neuropeptides, peptide hormones, antimicrobial peptides, and collagen fragments. Color intensity reflects modification frequency per class.
Label-Free Quantitation Reproducibility
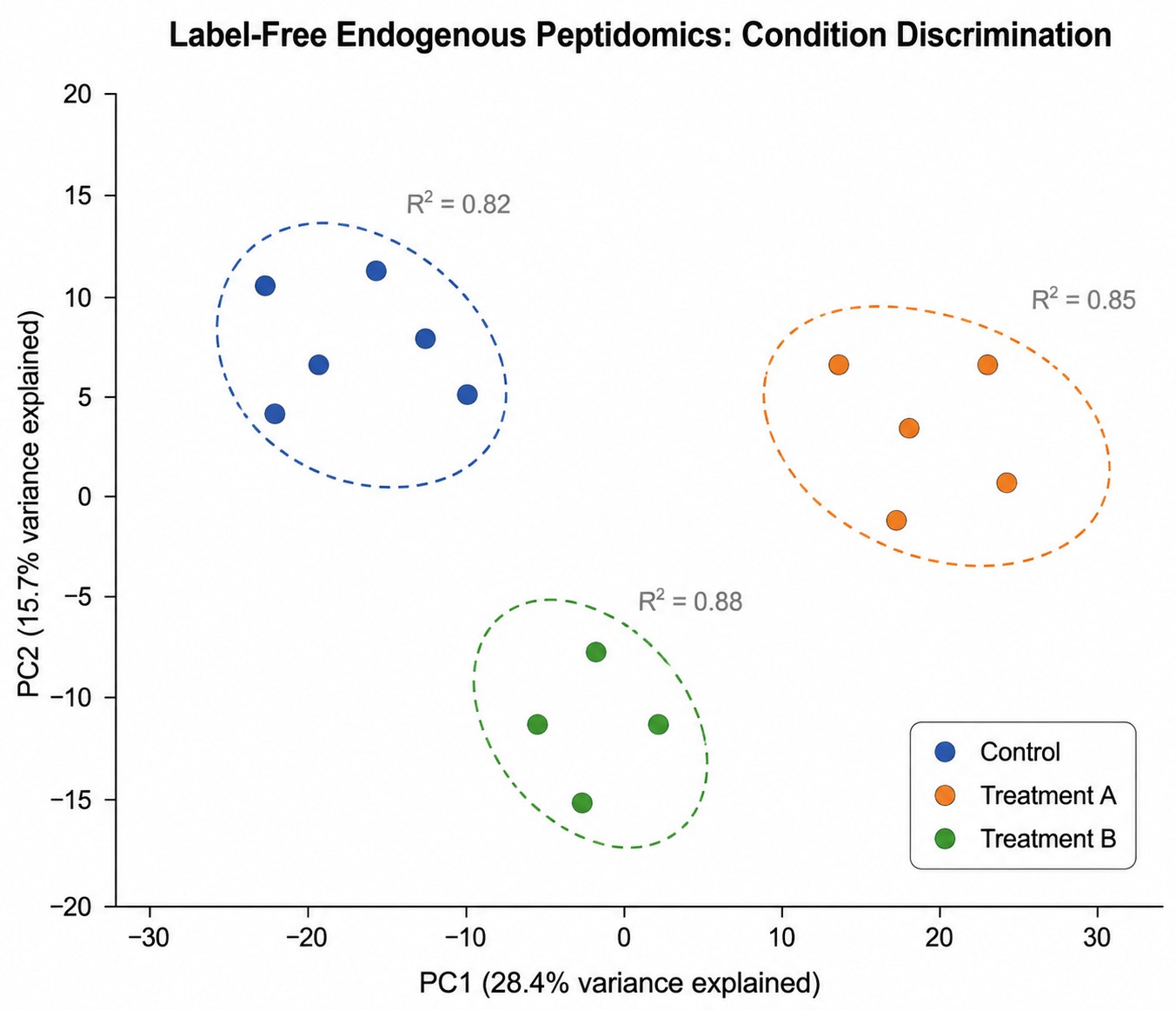
Principal component analysis (PCA) of label-free endogenous peptidomics data across three experimental groups. QC-normalized data shows tight intra-group clustering and clear inter-group separation along PC1 and PC2.
Protease Cleavage Site Preferences in Wound Fluid
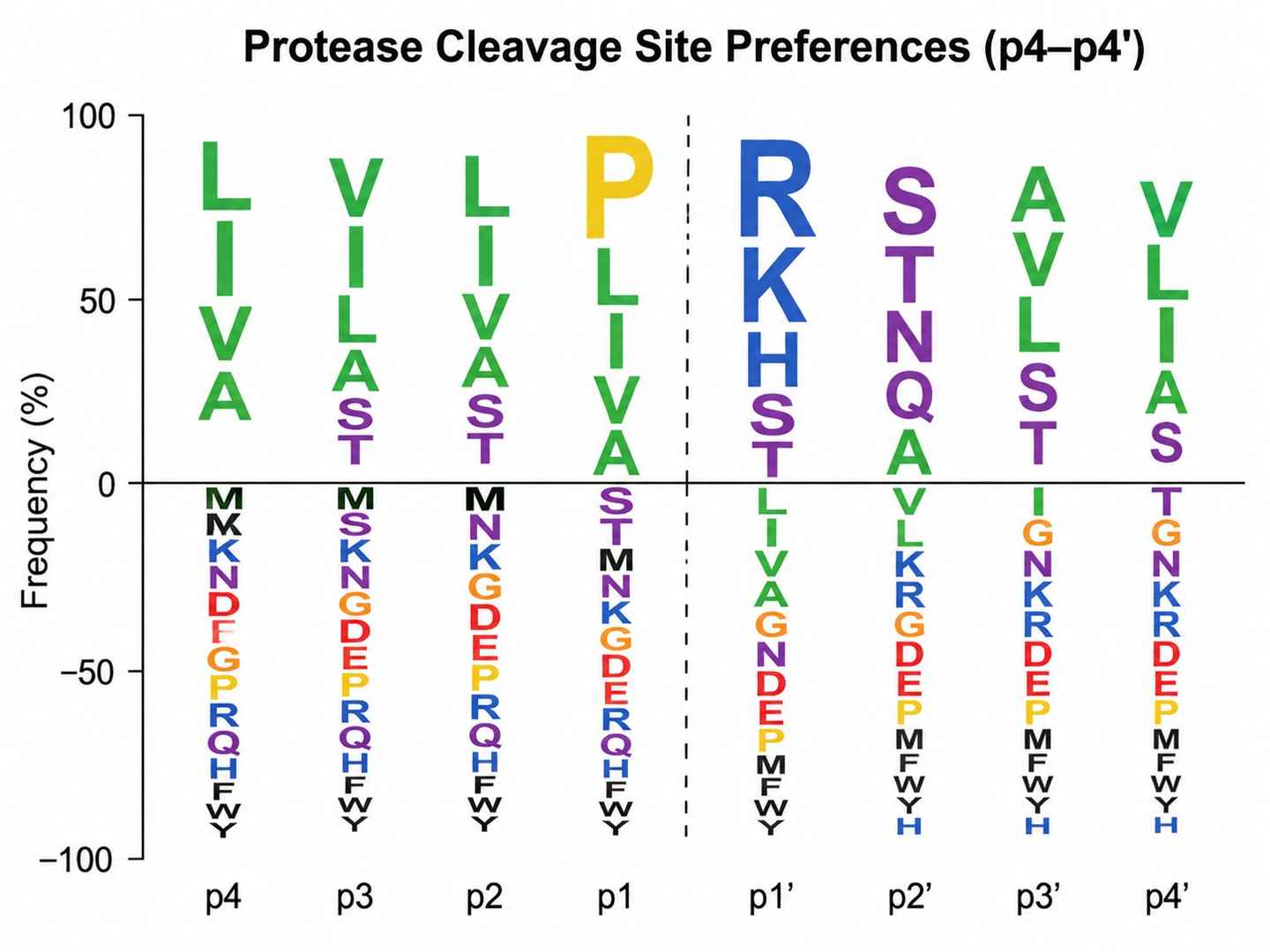
Protease cleavage site logo (p4–p4') derived from endogenous peptide sequences in wound fluid, colored by amino acid property (hydrophobic = green, acidic = red, basic = blue, polar = purple). Data from Hartman et al. 2024 Nature Communications, Figure 3.
Application Areas of Endogenous Peptidomics
- Biomarker Discovery — Identify endogenous peptide biomarkers for disease diagnosis, prognostic stratification, or treatment response prediction across multi-cohort biofluid collections — see also Immunopeptidomics Service.
- Neurobiology and Neuropeptide Research — Profile the functional neuropeptide repertoire from brain regions, CSF, or plasma to understand neural signaling dynamics under neurological or psychiatric disease conditions.
- Multi-Omics Integration — Combine endogenous peptidomics data with transcriptomics, proteomics, or metabolomics readouts to connect gene expression changes to the biologically active peptide products that drive cellular responses.
- Microbiome–Host Interaction Studies — Characterize host-derived antimicrobial peptides and protease-generated fragments in response to microbial colonization or infection.
- Nutrition and Food Science — Identify and quantify bioactive peptides derived from food proteins during gastrointestinal digestion or fermentation, supporting peptide-based nutraceutical development.
- Endocrine and Metabolic Disease Research — Quantify peptide hormones and their processing intermediates (e.g., GLP-1, insulin family peptides, adipokines) to study metabolic disorders, diabetes, and obesity.
- Toxicology and Natural Product Mechanism Studies — Profile endogenous peptide responses to xenobiotic exposure, venom components, or environmental toxins to elucidate mechanism-of-action and off-target effects.
- Tissue Remodeling and Wound Healing Biology — Monitor protease activity and collagen-derived peptide fragments during tissue repair, fibrosis, or pathological remodeling processes.
Endogenous Peptidomics Service Deliverables | What You Will Receive
- Raw LC-MS/MS Data Files (RAW Format)
Native instrument files for downstream reanalysis or method validation. - Complete Peptide Identification List and Sequence Information
Sequences, precursor masses, charge states, retention times, PTMs, and confidence scores for all identified endogenous peptides. - PTM Modification Annotation Table
Detailed annotation of C-terminal amidation, disulfide connectivity, N-terminal pyroglutamation, hydroxylation, bromination, oxidation, and phosphorylation with site localization. - Label-Free Quantitation Results with QC-Corrected Data
MS1 peak-area tables, normalized abundance values, EigenMS batch-corrected data, and statistical comparison results. - Statistical Visualizations (PCA, Volcano Plots, Clustering Heatmaps)
Publication-ready figures for condition-wise comparison and biological interpretation. - GO/KEGG Functional Enrichment Analysis
Biological process, cellular component, and molecular function annotations with pathway enrichment for source proteins of identified peptides. - Protease Cleavage Site Analysis
p4–p4' window frequency logos, protease activity inference, and comparison across conditions (where applicable). - Comprehensive PDF Analysis Report
Visual summary with peptide coverage statistics, PTM prevalence charts, quantitative comparisons, and representative MS/MS spectra.
Disclaimer: Creative Proteomics products and services are intended for Research Use Only (RUO). They are not intended for diagnostic, therapeutic, or clinical purposes.
What is the difference between endogenous peptidomics and conventional proteomics? +
Conventional proteomics digests proteins with trypsin before LC-MS/MS analysis, generating artificial peptide fragments that do not exist in living systems. Endogenous peptidomics analyzes naturally occurring, biologically active peptides that are produced by cellular proteolytic processing — without enzymatic digestion. This distinction is essential for studying neuropeptides, peptide hormones, antimicrobial peptides, and other bioactive fragments that are invisible in standard proteomic workflows.
What types of post-translational modifications can the platform detect? +
The platform detects and localizes a broad range of PTMs relevant to endogenous peptide function, including C-terminal amidation, N-terminal pyroglutamation, disulfide bond connectivity, hydroxylation, bromination, oxidation, and phosphorylation. PTM-aware search engines and fragmentation strategies (HCD, ETD, CID) are used to maximize modification detection confidence and site localization.
How much sample is required for endogenous peptidomics analysis? +
Minimum requirements are 50 µL of plasma, 10 mL of urine, 150 µL of cerebrospinal fluid, or 50 mg of tissue. Recommended amounts are 200 µL plasma, 50 mL urine, 500 µL CSF, or 200 mg tissue. Detailed requirements for all supported sample types are listed in the Sample Requirements section.
How does the platform prevent endogenous peptides from degrading during sample preparation? +
All sample processing is performed under acidic conditions (0.1–1% TFA or formic acid) with protease inhibitor cocktails immediately upon receipt or collection. Samples are kept cold throughout the extraction workflow, and peptide enrichment (30 kDa MWCO filtration + C18 SPE) is completed rapidly to minimize post-collection proteolysis. This preserves the authentic in-vivo peptide profile rather than generating artifactual cleavage products.
What bioinformatics analysis is included in the standard deliverables? +
Standard deliverables include the complete peptide identification list with PTM annotations, label-free quantitative tables with QC normalization and EigenMS batch correction, PCA / volcano plot / clustering heatmap visualizations, GO and KEGG functional enrichment for source proteins, protease cleavage site profiling (p4–p4' window analysis), and a comprehensive PDF summary report.
Peptide Clustering Enhances Large-Scale Analyses and Reveals Proteolytic Signatures in Mass Spectrometry Data
Journal: Nature Communications
Published: Volume 15, Article number 7128, 2024
Summary
Hartman and colleagues applied LC-MS/MS–based endogenous peptidomics to pig wound fluid and human chronic wound samples, identifying 14,950 endogenous peptide sequences from 539 source proteins. The study demonstrated that peptide clustering — grouping related peptide sequences into clusters based on shared precursor and cleavage characteristics — reduces dataset dimensionality by approximately 95%, dramatically decreases missing values (by 70±13%), and enables highly accurate classification of wound infection status (XGBoost accuracy: 94±2%). Critically, analysis revealed 68 statistically significant protease-activity clusters (33 proteins, Q<0.05), with 160 clusters uniquely associated with Pseudomonas aeruginosa–infected chronic wounds. These findings establish endogenous peptidomics as a powerful tool for classifying disease states and inferring active protease repertoires directly from mass spectrometry data.
Methods
The study characterized the endogenous peptide landscape in wound fluids from a pig burn-wound infection model and human chronic leg ulcers using LC-MS/MS. Peptide clustering was applied to group related peptide sequences, and the impact on dimensionality, missing values, and biological interpretability was systematically evaluated.
Key Analytical Features
- Sample types: Pig burn-wound fluid (n = multiple time points and infection conditions), human chronic leg ulcer wound fluid
- Identification: LC-MS/MS analysis and database search with post-translational modification annotation
- Clustering approach: Peptide sequence clustering based on shared precursor protein and cleavage site features to reduce dimensionality while preserving biological signal
- Validation: XGBoost machine learning classifier trained on clustered peptide data for infection-status classification
- Protease activity inference: p4–p4' cleavage site window analysis to identify protease-family–specific cleavage signatures across infection conditions
Results
Peptide Identification Coverage
- 14,950 endogenous peptides identified from 539 source proteins in pig wound fluid samples.
- Clustering reduced dataset dimensionality by approximately 95%, enabling practical downstream machine-learning analysis without sacrificing biological information.
- Missing value rate reduced by 70±13% through clustering-based imputation and signal consolidation.
Machine Learning Classification
- XGBoost classifier trained on clustered peptide data achieved 94±2% accuracy in discriminating infection status across wound samples.
- Cluster-based features provided superior classification performance compared with individual peptide markers, validating the biological relevance of the clustering strategy.
Protease Activity Signatures
- 68 statistically significant protease-activity clusters were identified across the dataset (33 source proteins, Q<0.05).
- 160 clusters were uniquely detected in Pseudomonas aeruginosa–infected chronic wound samples, defining a pathogen-specific proteolytic signature.
- Cut-site analysis (p4–p4' window) revealed distinct protease cleavage preferences between sterile and pathogen-colonized wound environments.
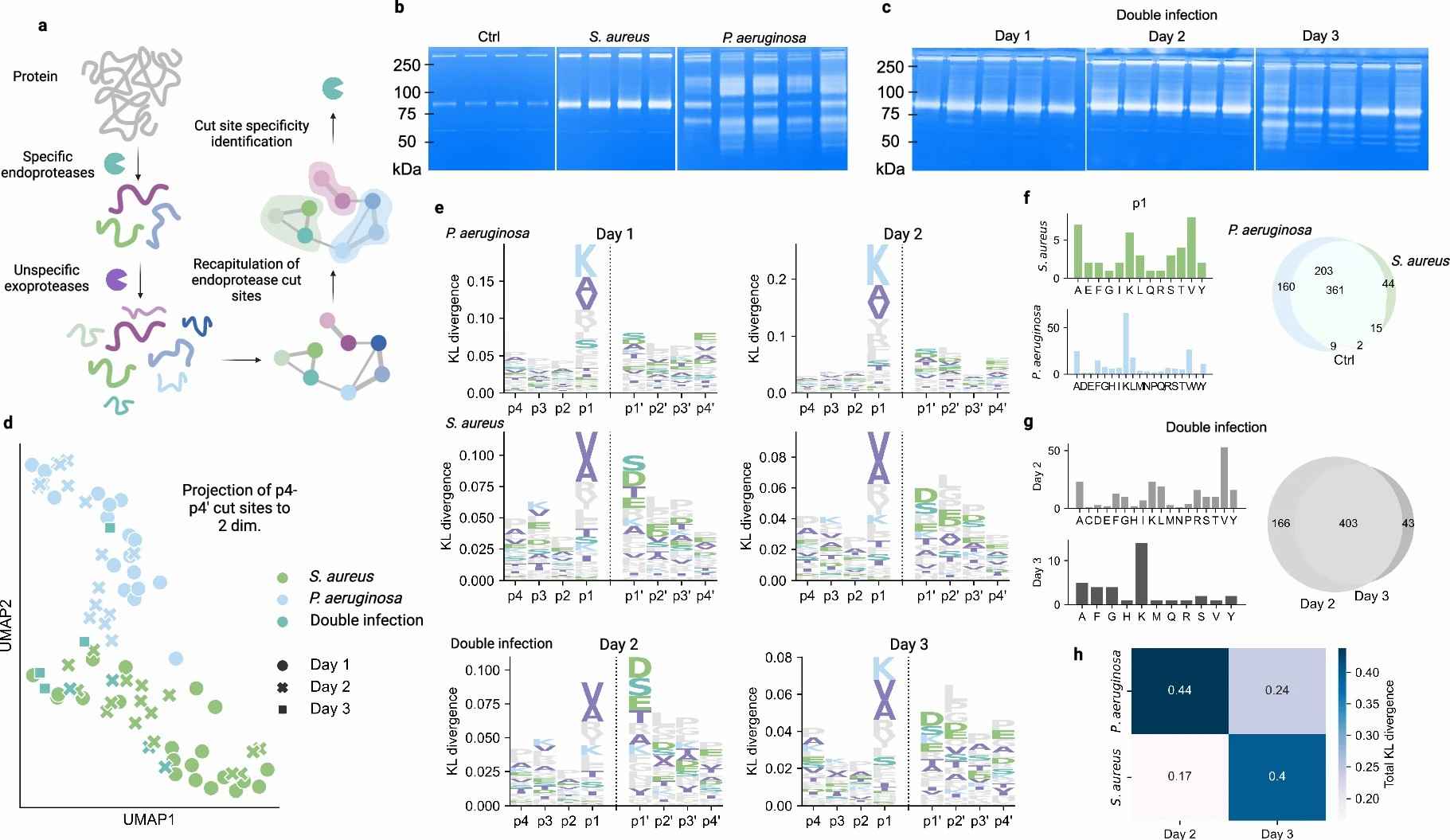
Figure 3. Cut-site analysis reveals pathogen-specific protease patterns in wound fluid. Logoplots show amino acid frequency at positions p4–p4' flanking identified endogenous peptide cleavage sites, comparing sterile wound, S. aureus–infected, and P. aeruginosa–infected conditions. From Hartman et al. 2024 Nature Communications, Figure 3.
What This Means for Your Endogenous Peptidomics Project
- The 94% infection-classification accuracy achieved with clustered peptide features demonstrates that LC-MS/MS–based endogenous peptidomics data can reliably discriminate between distinct biological or disease states — supporting its use for biomarker discovery and diagnostic development.
- The ~95% dimensionality reduction achieved by peptide clustering resolves a fundamental challenge in endogenous peptidomics data analysis: managing thousands of individual peptide variables. This approach is directly applicable to any multi-condition comparative peptidomics study.
- The identification of 160 P. aeruginosa–specific protease-activity clusters shows that endogenous peptide cleavage signatures encode sufficient pathogen-identity information to distinguish infection types. This has direct implications for clinical wound-management and infectious disease research.
- Cut-site profiling (p4–p4' window analysis) enables inference of the active protease repertoire from endogenous peptide sequences alone, without requiring direct protease activity assays. This is a powerful tool for studying inflammatory cascades, tissue remodeling, and host–pathogen interactions.
Reference
- Hartman E, Forsberg F, Kjellström S, Petrlova J, Luo C, Scott A, Puthia M, Malmström J, Schmidtchen A. (2024). Peptide clustering enhances large-scale analyses and reveals proteolytic signatures in mass spectrometry data. Nature Communications, 15, 7128. https://doi.org/10.1038/s41467-024-51589-y