Why Neoantigen Prediction Alone Is Not Enough
In modern immuno-oncology, next-generation sequencing (NGS) allows for the rapid identification of thousands of tumor-specific somatic mutations. Standard bioinformatic pipelines process these mutations through baseline algorithms to predict which mutated peptides might bind to patient-specific HLA molecules. However, this raw prediction phase routinely generates an overwhelming list of theoretical candidates—often thousands of peptides long—with a false-positive rate that can exceed 90%.
For a candidate to be truly actionable, it must not only possess theoretical binding affinity but also be actively transcribed, properly cleaved by the proteasome, successfully transported, and uniquely immunogenic. Filtering this massive dataset requires specialized bioinformatics expertise. Serving as a crucial bridge within our broader neoantigen discovery service, our prediction and prioritization service applies multi-layered, evidence-aware ranking logic to distill thousands of raw predictions down to a high-confidence, testable shortlist.
What Is the Difference Between Prediction, Prioritization, and Validation?
Navigating the neoantigen discovery pipeline requires distinguishing between three distinct phases of data confidence:
- Prediction: The automated, algorithmic process of guessing theoretical HLA binding affinity directly from raw genomic variations. This yields high volume but low clinical confidence.
- Prioritization: The advanced computational ranking of predicted candidates. It integrates transcriptomic expression (RNA-seq), proteasomal processing algorithms, clonality, and AI-driven immunogenicity scoring to objectively rank the candidates most likely to succeed in functional assays.
- Validation: The physical wet-lab confirmation of the computational shortlist. This typically involves transitioning the prioritized candidates to our mass spectrometry-based neoantigen discovery service for absolute experimental proof of biological presentation.
Key Application Areas
Our prioritization services are designed to support critical decision-making in highly targeted translational oncology pipelines:
Personalized Cancer Vaccine Target Selection
Safely select the top 10 to 20 highly specific, patient-restricted neoepitopes required for formulating potent mRNA or synthetic long peptide (SLP) vaccines.

TCR or T-Cell Therapy Target Prioritization
Identify and rank tumor-restricted targets that exhibit strong T-cell receptor recognition potential, effectively minimizing the risk of on-target, off-tumor toxicities.
Biomarker Discovery for Immunotherapies
Profile and rank the neoantigen burden across patient cohorts to uncover mechanisms of immune evasion and identify predictive biomarkers for immune checkpoint blockade response.
Tumor-Infiltrating Lymphocyte (TIL) Expansion
Prioritize the most immunogenic mutated peptides to selectively stimulate and expand reactive CD8+ and CD4+ TILs ex vivo for adoptive cell transfer.
Shared Neoantigen Screening
Screen across large patient cohorts to identify and rank high-frequency, shared driver mutations suitable for "off-the-shelf" broad-spectrum therapeutic development.
Pre-Clinical Murine Model Profiling
Apply our advanced ranking algorithms to syngeneic mouse models (e.g., B16F10, CT26) to robustly validate target immunogenicity prior to human clinical trials.
What We Offer in Neoantigen Prediction & Prioritization
We provide a comprehensive computational suite that moves beyond simple affinity scoring, offering a transparent and highly tailored approach to candidate generation and ranking.
AI-Driven Neoantigen Prediction and Prioritization
To overcome the limitations of basic algorithmic filtering, Creative Proteomics employs an advanced hybrid bioinformatics strategy. We utilize industry-standard tools as foundational layers and enhance them with proprietary AI-driven prioritization logic.
Our AI-assisted ranking focuses on two critical technical pillars:
- Presentation Prediction Trained on MS Data: Our machine learning models are continuously refined using large-scale, high-resolution HLA peptidomics analysis datasets. This allows the AI to recognize native processing motifs, accurately predicting proteasomal cleavage efficiency and TAP transport likelihood—factors that standard affinity algorithms ignore.
- Structural Immunogenicity Modeling: A peptide may bind perfectly to an HLA molecule but still fail to elicit a T-cell response. We apply AI to 3D structural modeling (MHC-peptide-TCR docking simulations) to assess the "foreignness" of the exposed peptide surface, prioritizing candidates with the highest probability of triggering robust T-cell recognition.
*Note: While AI drastically improves shortlist accuracy, computational prioritization remains a predictive science; top-ranked candidates must still undergo experimental validation.
Candidate Types We Can Prioritize
Our bioinformatics pipelines are equipped to handle a diverse array of tumor-specific variations, ensuring comprehensive profiling of the potential immunopeptidome.
| Antigen Type | Genomic Origin | Prioritization Strategy |
|---|---|---|
| SNV-Derived Neoantigens | Point mutations in coding exons. | Ranking based on differential binding affinity (Mutant vs. Wild-type) and RNA expression. |
| Indel-Derived Neoantigens | Frameshift mutations altering reading frames. | Generation of novel downstream peptide sequence windows for comprehensive HLA binding prediction. |
| Fusion-Derived Neoantigens | Chromosomal translocations / chimeric transcripts. | Isolation of junction-spanning peptides and evaluation of junctional immunogenicity. |
| Splice-Derived Neoantigens | Intron-retention or exon-skipping events. | RNA-seq guided mapping to predict novel open reading frames and unique presentation. |
| Cryptic / Non-Canonical | Translation of UTRs or non-coding RNAs. | 6-frame translation integration and prioritization based on tumor-exclusive expression. |
| TAAs & Oncoviral Antigens | Overexpressed normal genes or viral integration. | Baseline screening for common shared antigens to supplement personalized vaccine pipelines. |
How Our Prioritization Workflow Works
Our computational workflow operates through a structured, multi-parametric filtration process designed to ensure data transparency at every stage.
Project Intake & Data Review
Define optimal pipeline parameters
Variant Calling & HLA Mapping
Identify somatic mutations & alleles
Primary Candidate Generation
Baseline predictive algorithms
AI & Multi-Factor Prioritization
Filter & rank based on RNA & TCR
Shortlist Reporting
Generate highly actionable shortlist
1
Project Intake & Data Review
We systematically review your provided genomic data (FastQ/BAM/VCF), RNA-seq files, and clinically derived HLA typing data to define the optimal pipeline parameters.
2
Variant Calling & HLA Mapping
Somatic mutations are identified against matched normal data, and the patient's HLA Class I and/or Class II alleles are computationally confirmed or directly inputted.
3
Primary Candidate Generation
Baseline predictive algorithms are deployed to generate a comprehensive list of all possible mutated peptides capable of anchoring into the patient's HLA grooves.
4
AI & Multi-Factor Prioritization
Candidates are aggressively filtered and ranked based on RNA expression (TPM), predicted proteasomal cleavage, TCR recognition potential, and tumor clonality.
5
Shortlist Reporting
A finalized, highly actionable shortlist (e.g., top 20 candidates) is generated, accompanied by a detailed prioritization rationale report to guide your next experimental steps.
Prediction-Only vs. Prioritization-Driven Strategy
Understanding the difference in service tiers helps align our bioinformatics output with your project's current phase and budget.
| Dimension | Standard Prediction Only | Prioritization-Driven Ranking (This Service) |
|---|---|---|
| Candidate Volume | Thousands of theoretical hits. | A refined shortlist (e.g., Top 10-50). |
| Evidence Depth | Genomic mutation + Binding %Rank. | Genomics + Transcriptomics + Processing + Immunogenicity. |
| False-Positive Control | Very Low (>90% false positives). | High (Strict multi-factor filtration). |
| Downstream Usability | Requires extensive internal bioinformatics work. | Ready for immediate peptide synthesis or MS validation. |
| Best-Fit Project Stage | Early exploratory screening / TMB estimation. | Vaccine formulation / TCR target selection. |
Sample and Data Requirements
To perform highly accurate neoantigen prioritization, matched genomic and transcriptomic data are heavily preferred.
| Input Type | Typical Format | Requirement Status | Why It Matters |
|---|---|---|---|
| Tumor DNA / WES / WGS | FastQ, BAM, or VCF | Required | Provides the foundational raw somatic mutations (SNVs, indels) driving neoantigen creation. |
| Matched Normal DNA | FastQ, BAM, or VCF | Required | Essential for accurately filtering out patient-specific germline variations to isolate true tumor mutations. |
| RNA-seq Data | FastQ or BAM | Highly Recommended | Proves the mutated gene is actively transcribed; dramatically reduces false positives. |
| HLA Typing Data | Text / Clinical Report | Required | Determines the precise HLA allotypes (e.g., HLA-A*02:01) to which the peptides must bind. (Can be computationally inferred if raw WES is provided). |
| Optional: Proteogenomics Inputs | MS RAW files | Optional | If transitioning to our proteogenomics analysis service, physical mass spectrometry data is required for ultimate validation. |
From Raw Data to Actionable Targets: Prioritization Outputs
Our deliverables include intuitive, publication-ready data visualizations that demystify the AI-driven ranking logic and clearly highlight your top candidates.
Candidate Ranking Matrix
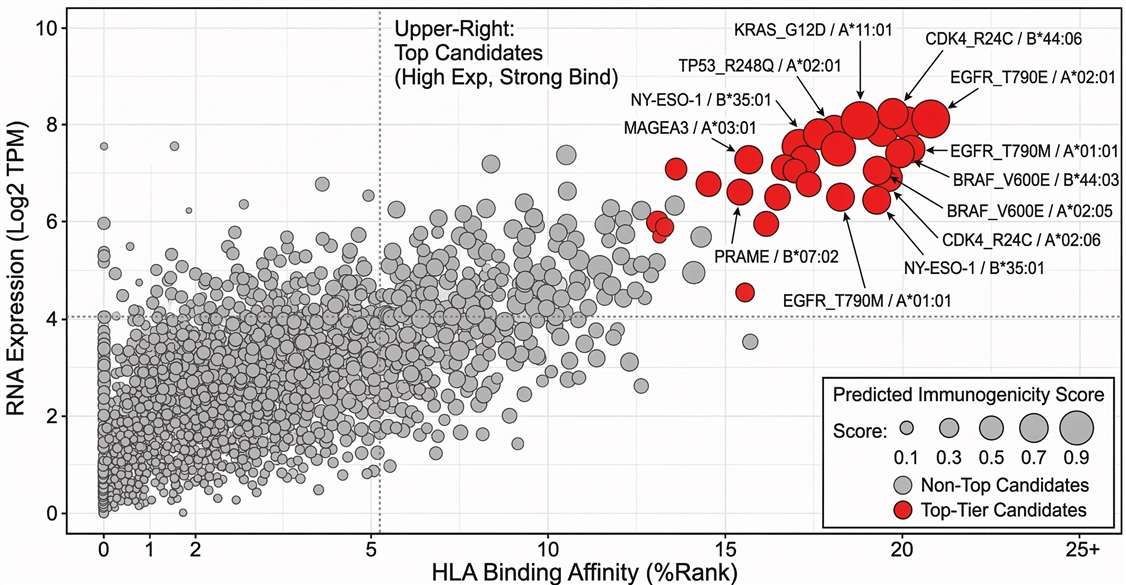
Multi-parameter neoantigen ranking matrix identifying high-confidence, immunogenic targets for downstream validation.
Binding Affinity Distribution
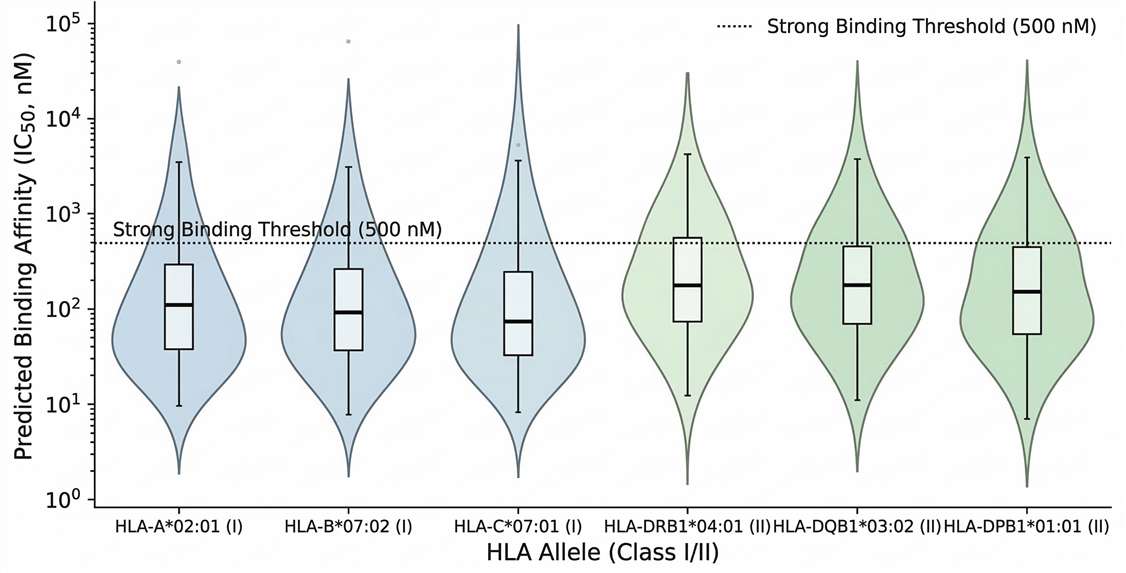
Distribution of predicted neoepitope binding affinities across distinct patient HLA class I and II alleles.
Variant Class Composition
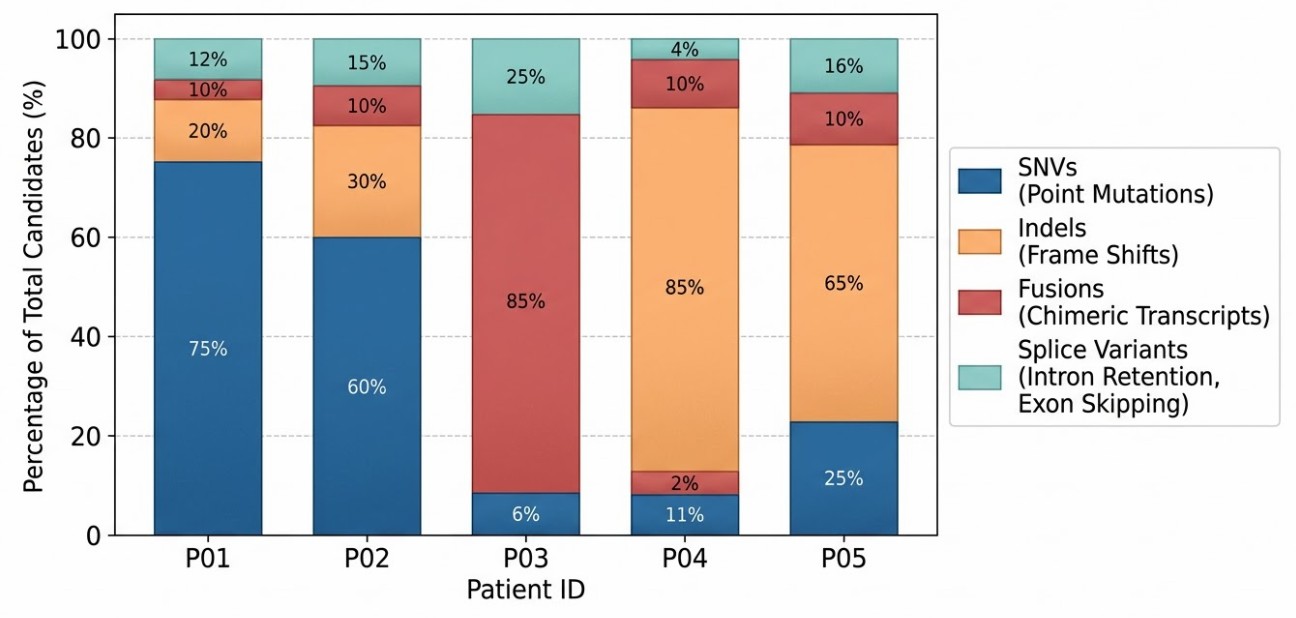
Variant class composition of the prioritized neoantigen shortlist, encompassing complex structural and cryptic variants.
Benchmarking: Prioritization Pipeline Performance
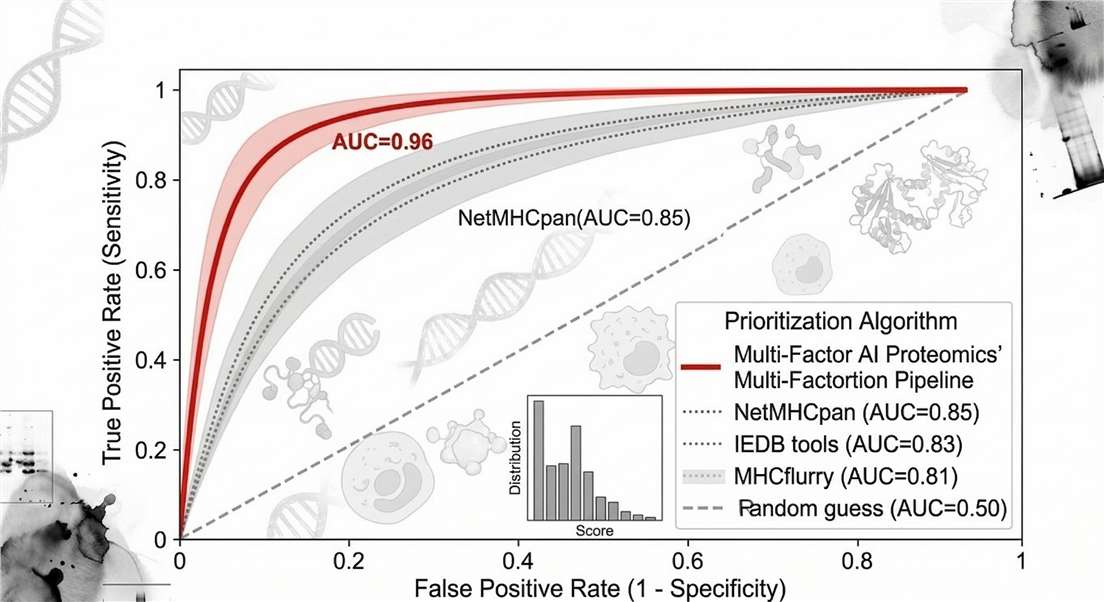
ROC curve demonstrating Creative Proteomics's Multi-Factor AI pipeline significantly outperforming standard single-metric algorithms in identifying presented neoantigens.
Deliverables | What You Will Receive
Our output packages are rigorously formatted to seamlessly integrate with your downstream experimental workflows. Upon completion, we provide a smooth transition path to tumor neoantigen discovery and validation services for physical confirmation.
- Ranked Neoantigen Candidate Table: A definitive Excel/CSV matrix of the top candidates, sorted by our proprietary multi-factor AI priority score.
- Candidate Annotation Sheet: Detailed genomic context for every shortlisted peptide, including gene source, variant type, wild-type sequence comparison, and chromosomal coordinates.
- HLA Binding & Expression Matrix: The raw computational values supporting the ranking, including precise NetMHCpan %Rank scores and RNA-seq TPM values.
- Prioritization Rationale Report: A detailed methodology document outlining the exact thresholding logic, AI parameters, and decoy strategies used to refine the list.
- Executive Summary PDF: A high-level visual summary featuring all demo result graphics, ideal for team presentations and regulatory documentation.
References
- pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunology Research, 2020. https://doi.org/10.1158/2326-6066.CIR-19-0401
- Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell, 2020. https://doi.org/10.1016/j.cell.2020.09.015
What data are required for neoantigen prediction and prioritization? +
At minimum, we require patient-specific tumor DNA sequencing data (WES/WGS) and matched normal DNA to identify true somatic mutations. For accurate prioritization and false-positive reduction, matched RNA-seq data is highly recommended to confirm active gene expression.
Is HLA typing necessary? +
Yes, high-resolution HLA typing (Class I and/or Class II) is required because prediction algorithms calculate binding against specific allele structures. If you do not have clinical HLA reports, we can computationally infer the HLA haplotype from your raw sequencing data.
How is prioritization different from prediction alone? +
Prediction simply calculates whether a mutated peptide could theoretically bind to an HLA molecule based on its sequence. Prioritization adds critical biological context—such as whether the gene is expressed, whether the cell's machinery will properly cut the peptide, and whether a T-cell is likely to recognize it—to rank the most viable targets.
Can fusion and splice-derived candidates be included? +
Yes. Unlike basic pipelines that only look at SNVs, our advanced algorithms utilize your RNA-seq data to map complex structural variants, allowing us to prioritize highly immunogenic fusion-derived and splice-variant neoantigens.
Do you support Class I and Class II prioritization? +
Yes. We can generate and rank candidates for both CD8+ T-cell targets (HLA Class I) and CD4+ T-cell targets (HLA Class II), providing comprehensive coverage of the tumor's immunological potential.
How do you reduce false positives? +
We reduce false positives by shifting from single-metric reliance to multi-factor integration. Candidates are eliminated if they fail to meet RNA expression thresholds, show poor predicted proteasomal cleavage, or exhibit sequences likely to induce central tolerance.
Can prioritized candidates be forwarded to MS or proteogenomics validation? +
Absolutely. Computational prioritization is the optimal first step. Once the shortlist is generated, we highly recommend transitioning the targets to our immunopeptidomics service or targeted MS platforms for definitive physical validation of presentation.
Case Study: Prioritization of Cancer Therapeutic Neoantigens Through Machine Learning
Journal: BMC Medical Genomics
Published: Volume 11, 2018
Summary
While predicting MHC-peptide binding is a critical first step, high false-positive rates inherently hinder the clinical application of neoantigens. Researchers recognized that an ideal prioritization pipeline must look beyond single-metric affinity scoring and incorporate multiple biological features—such as expression levels, cleavage probability, and TCR recognition potential—to successfully identify true immunogenic targets. This study proved that machine learning models successfully identify true immunogenic neoantigens that have been experimentally validated, effectively isolating the "needle in the haystack."
Methods
This published study introduces a machine learning-based neoantigen prioritization program. The computational pipeline extracted somatic mutations and RNA expression data, generating candidate peptides.
Key Technical Features:
- Multi-Factor Training: Instead of relying solely on IC50 binding scores, the machine learning model was trained using nine distinct immunogenicity features.
- Biological Context Integration: These features included agretopicity (MHC binding), epitopicity (TCR recognition), RNA abundance, and proteasomal cleavage prediction algorithms.
- Validation: Candidates were validated against independent melanoma and leukemia cohorts.
Creative Proteomics can offer advanced bioinformatics services that mirror and support this research pipeline, including:
- Multi-parametric neoantigen filtering and candidate ranking.
- Integration of WES and RNA-seq data to support AI-driven immunogenicity scoring.
- Seamless transition to MS-based immunopeptidomics for physical target validation.
Results
Multi-Factor Modeling Outperforms Single-Metric Scoring
- The multi-factor machine learning model demonstrated robust performance, significantly outperforming traditional, single-metric ranking approaches (like isolated binding affinity).
Identification of Validated Immunogenic Targets
- By mathematically weighting expression data alongside structural and cleavage predictions, the pipeline successfully ranked and identified clinically viable neoantigen candidates that trigger T-cell responses.
Reference
- Prioritization of cancer therapeutic neoantigens through machine learning approaches. BMC Medical Genomics, 2018. https://doi.org/10.1186/s12920-018-0405-3
For Research Use Only. Not for use in diagnostic procedures.