Degradomics for Protease Substrate Discovery
Degradomics is a specialized branch of proteomics dedicated to the system-wide analysis of proteolysis. Unlike traditional global profiling that measures total protein abundance, degradomics is a protease-focused mass spectrometry workflow centered on identifying exact cleavage events, discovering physiological protease substrates, and extracting mechanistic insights. By isolating and sequencing cleavage-derived neo-termini, researchers can uncover how specific proteases drive biological pathways, regulate protein half-lives, and generate bioactive fragments in both healthy and disease states.
Protease Profiling and Cleavage Site Mapping
Protease substrate profiling goes beyond identifying which enzymes are present; it reveals what those enzymes are actively doing. Through precise cleavage-site mapping, we identify the specific peptide bonds broken by a protease of interest. This relies on the enrichment and detection of neo-termini—the newly created N- or C-terminal sequences generated upon proteolytic cleavage. By sequencing these cleavage-derived peptides using advanced tandem mass spectrometry, we map the exact cleavage positions back to the parent protein sequence, confirming direct substrate interactions and defining the consensus recognition motifs unique to your target protease.
Applications in Biomarker Discovery and Mechanism Research
Proteolytic cleavage is an irreversible post-translational modification crucial to numerous physiological processes. Our degradomics service supports critical research applications across multiple disciplines:
Biomarker Discovery
Identifying stable cleavage fragments circulating in biofluids during disease progression, particularly in oncology and inflammation.
Inflammation & Infection
Profiling host-pathogen extracellular proteolysis, viral processing, and immune signaling cascades.
Target Validation
Confirming that a therapeutic protease inhibitor effectively blocks substrate cleavage in complex biological matrices.
Mechanism Research
Elucidating protease-mediated pathways, from the shedding of cell-surface receptors to the activation of dormant pro-hormones.
What We Offer in Degradomics and Protease Profiling
Cleavage-Derived Outputs and Source Types
This specialized workflow generates specific output classes designed to characterize proteolytic networks deeply:
| Output Type | Description | Biological Value |
|---|---|---|
| Cleavage-Derived Peptides | Non-tryptic fragments identified via mass spectrometry matching. | Confirms that localized protein degradation or processing is actively occurring. |
| Neo-N Termini | Newly exposed protein N-termini resulting from a specific cleavage event. | Allows for precise base-pair mapping of the exact cleavage site driven by the protease. |
| Substrate Candidates | Parent proteins matched to highly enriched cleavage-derived peptides. | Identifies which biological targets are actively regulated by the protease network. |
| Comparative Cleavage Events | Differential abundance of cleavage fragments across sample conditions. | Highlights how a disease state, treatment, or protease addition alters global proteolysis. |
Advantages of Degradomics for Protease Research
Standard mass spectrometry measures intact or trypsin-digested proteins, often masking the critical functional fragments generated by endogenous enzymes. Degradomics provides distinct advantages for protease biology:
Technology Platform for Degradomics Analysis
Our mass spectrometry-centered workflow is specifically optimized for peptide-level resolution and terminomics. Utilizing high-resolution LC-MS/MS platforms, we capture and sequence low-abundance functional fragments that are typically lost in standard proteomics. For specialized enrichment strategies, we employ approaches compatible with N-terminomics and selective peptide capture.
To meet the stringent analytical demands of protease substrate discovery, our laboratory is equipped with industry-leading mass spectrometry systems designed for high sensitivity and precise cleavage-site mapping:
| Instrument Platform | Key Analytical Specifications | Advantage for Degradomics |
|---|---|---|
| Orbitrap High-Resolution MS (e.g., Orbitrap Exploris 480 / Eclipse) | Resolution: Up to 480,000 at m/z 200 Mass Accuracy: < 1 ppm (internal) Scan Speed: Up to 40 Hz |
Ultra-high mass accuracy ensures confident identification of non-tryptic cleavage-derived peptides and isobaric post-translational modifications. |
| Ion Mobility Mass Spectrometry (e.g., timsTOF Pro 2) | Acquisition: PASEF (>100 Hz) Resolution: > 60,000 Dimension: 4D-Proteomics (CCS) |
Enhanced sensitivity and orthogonal separation of co-eluting isomeric peptides, critical for detecting low-abundance neo-termini in complex tissue lysates. |
| Nano-Flow LC Systems (e.g., nanoElute, UltiMate 3000 RSLCnano) | Flow Rate: ~200–300 nL/min Gradient: High-precision reproducibility |
Maximizes peptide ionization efficiency and retention time stability, ensuring robust quantitative accuracy for comparative degradomics across large sample cohorts. |
This robust analytical foundation is similar to the precision required in Immune Peptide Mass Spectrometry Analysis, ensuring high sensitivity for non-tryptic, biologically generated peptides. Whether you require global comparative profiling or targeted substrate validation, our platform integrates advanced bioinformatics to deliver mechanism-focused biological interpretations. Researchers requiring highly specific peptide characterization across varying applications may also explore our adjacent Immunopeptidomics Service.
Workflow for Degradomics and Protease Profiling
Project Design & Consultation
Treated-vs-control models
Sample Review & Preparation
Extraction & stabilization
Enrichment Strategy (Optional)
N-terminal labeling & isolation
LC-MS/MS Analysis
High-resolution tandem MS
Cleavage Calling & Annotation
Bioinformatics alignment
QC Review & Pathway Mapping
Statistical & functional validation
1
Project Design & Consultation
We collaborate to establish treated-versus-control models or in vitro protease incubation strategies optimized for robust cleavage detection.
2
Sample Review & Preparation
Biological samples undergo rigorous quality control. Proteins are extracted and stabilized rapidly to prevent unwanted background degradation before controlled proteolysis or enrichment.
3
Enrichment Strategy (Optional)
For targeted terminomics, samples may undergo N-terminal labeling and negative selection to isolate neo-termini, drastically reducing the complexity of the internal tryptic background.
4
LC-MS/MS Analysis
High-resolution tandem mass spectrometry identifies and quantifies the peptide fragments, carefully distinguishing biologically relevant cleavage events from routine sample processing artifacts.
5
Cleavage Calling & Annotation
Specialized bioinformatics pipelines align detected non-tryptic peptides against the host proteome, mapping exact cleavage sites and identifying candidate parent substrates.
6
QC Review & Pathway Mapping
Data is filtered through strict statistical thresholds. Validated substrates are then mapped to biological pathways, delivering a clear view of systemic protease activity.
Degradomics vs Standard Proteomics
Choosing the right analytical approach is critical for studying protease biology. The table below clarifies when degradomics is the superior choice for mechanistic research.
| Dimension | Standard Global Proteomics | Degradomics & Protease Profiling | Targeted Cleavage Assay |
|---|---|---|---|
| Primary Question Answered | Which proteins changed in total abundance? | Where was the protein cleaved, and what are the substrates? | Is this specific cleavage fragment present in my sample? |
| Main Output | Total protein fold-change lists. | Cleavage-site maps and substrate candidate tables. | Absolute quantification of a known neo-peptide. |
| Strength | Comprehensive overview of cellular expression states. | Identifies exact processing events and active proteolysis networks. | High throughput and high sensitivity for validated targets. |
| Limitation | Masks specific cleavage events due to heavy tryptic digestion. | Requires rigorous comparative design to filter background noise. | Cannot discover new substrates or unknown cleavage sites. |
| Best-Fit Project Stage | Early exploratory expression profiling. | Substrate discovery and mechanism elucidation. | Downstream validation and biomarker screening. |
Solution Selection Strategy:
- Choose degradomics when the main goal is cleavage-site mapping or protease substrate discovery.
- Choose standard proteomics when the main goal is simply global protein abundance change.
- Choose comparative degradomics when treated-vs-control proteolysis differences are biologically important.
- Add targeted follow-up assays when a shortlist of validated cleavage events needs orthogonal confirmation.
Sample Requirements for Degradomics Studies
We support both in vitro substrate discovery assays and in vivo comparative profiling. Proper sample handling is crucial to prevent artifactual proteolysis.
| Sample Type | Typical Input | Required or Optional | Best Use | Notes |
|---|---|---|---|---|
| Cell Lysates | Untreated and protease-perturbed samples | Optional | Comparative intracellular proteolysis | Paired treated-vs-control design strongly preferred. Avoid harsh detergents like SDS unless previously cleared. |
| Conditioned Media | Secreted protein fraction | Optional | Extracellular protease activity studies | Clarify collection conditions and use serum-free media if possible to reduce background. |
| Tissue Lysates | Matched biological samples | Optional | Disease vs. control cleavage profiling | Immediate snap-freezing or rapid heat-stabilization is critical to preserve the endogenous degradome. |
| Recombinant Substrates | Defined protein substrate system | Optional | Protease-of-interest cleavage mapping | Sequence and treatment design required. Highly efficient for mapping exact consensus motifs. |
| In Vitro Protease-Treated Samples | Control vs. treated material | Optional | Direct substrate discovery in complex lysates | Incubation design (enzyme-to-substrate ratio, buffer, time) should be tightly specified to avoid over-digestion. |
(Note: Exact input amounts, enrichment yields, and replicate expectations should be confirmed case by case through project consultation.)
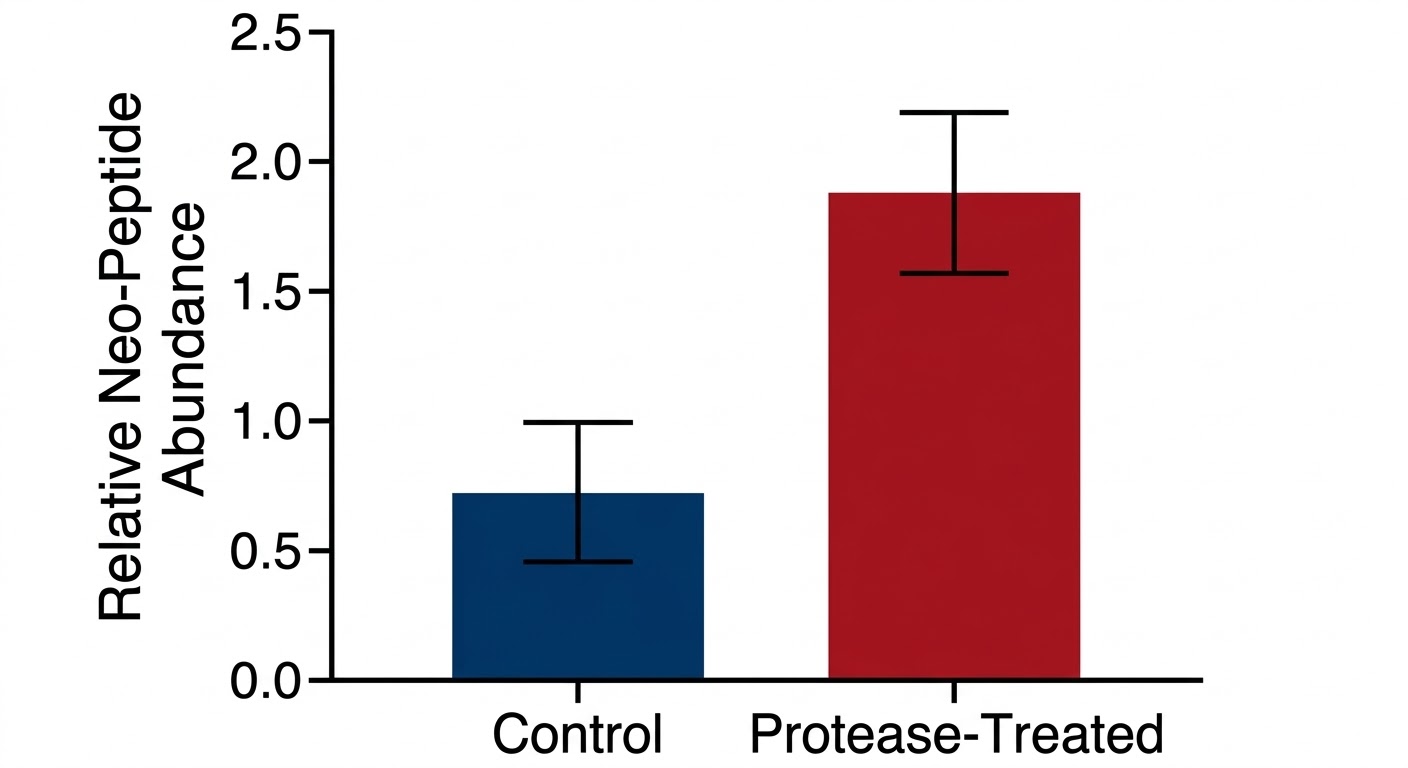
Example Results from Degradomics Analysis
Our bioinformatics team processes complex mass spectrometry data into highly interpretable, publication-grade scientific formats to clearly answer where cleavage occurred and which pathways are affected.
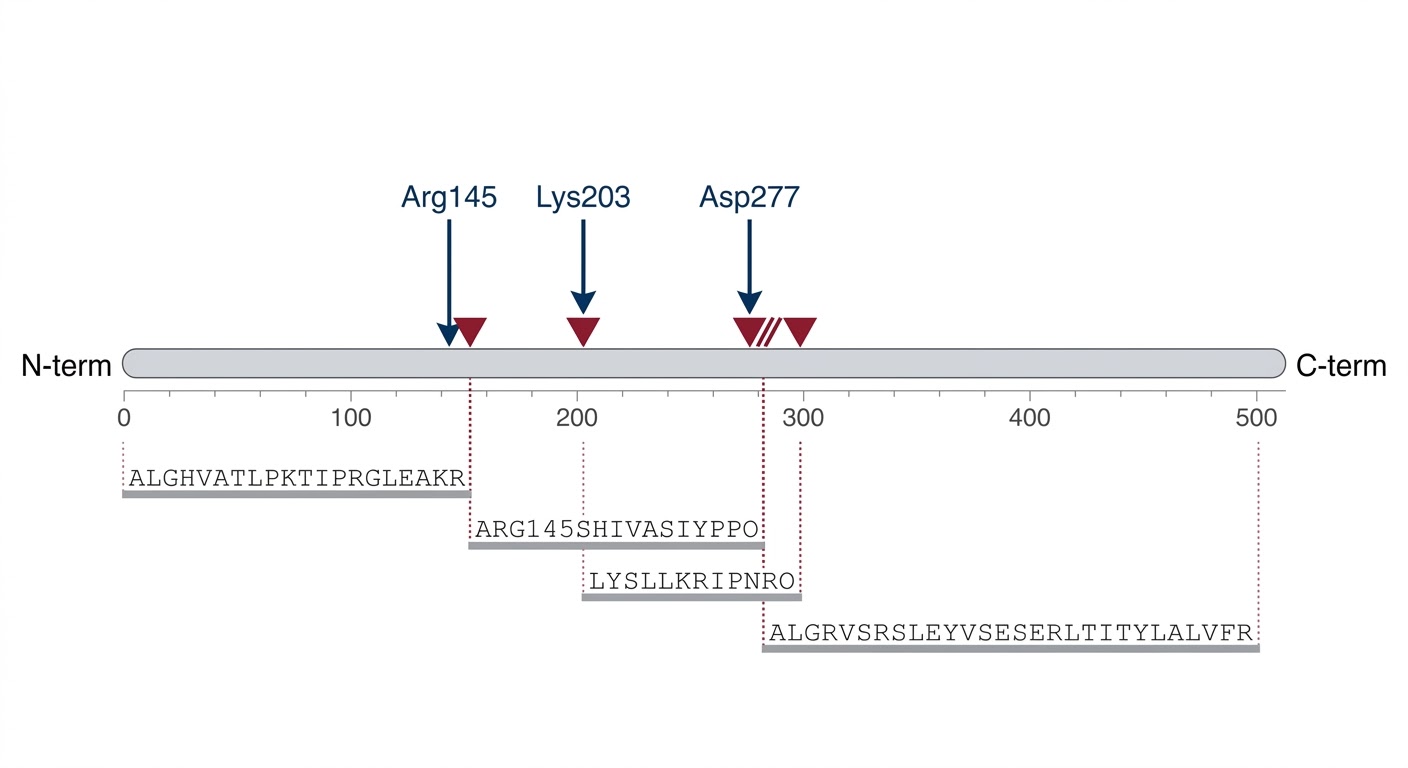
High-Resolution Cleavage-Site Map
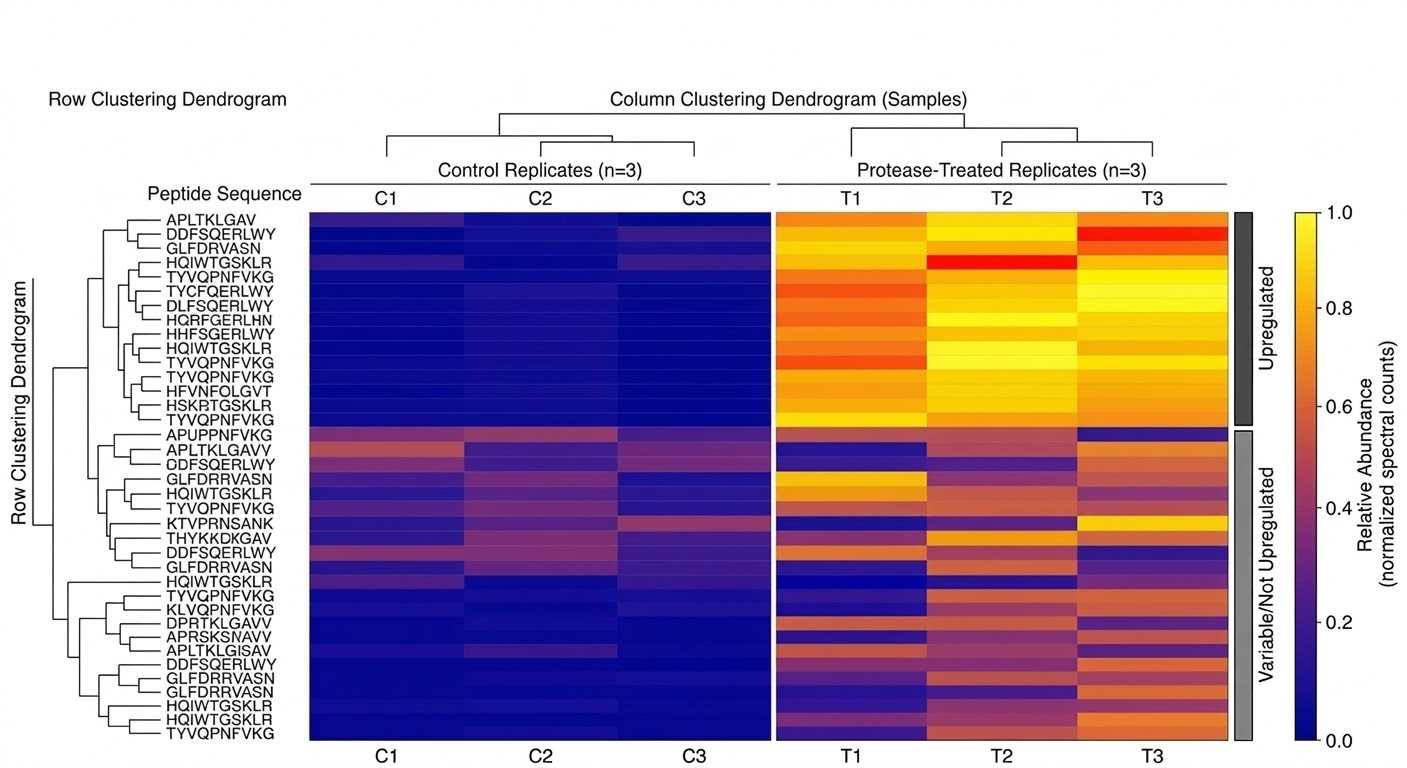
Comparative Proteolysis Heatmap
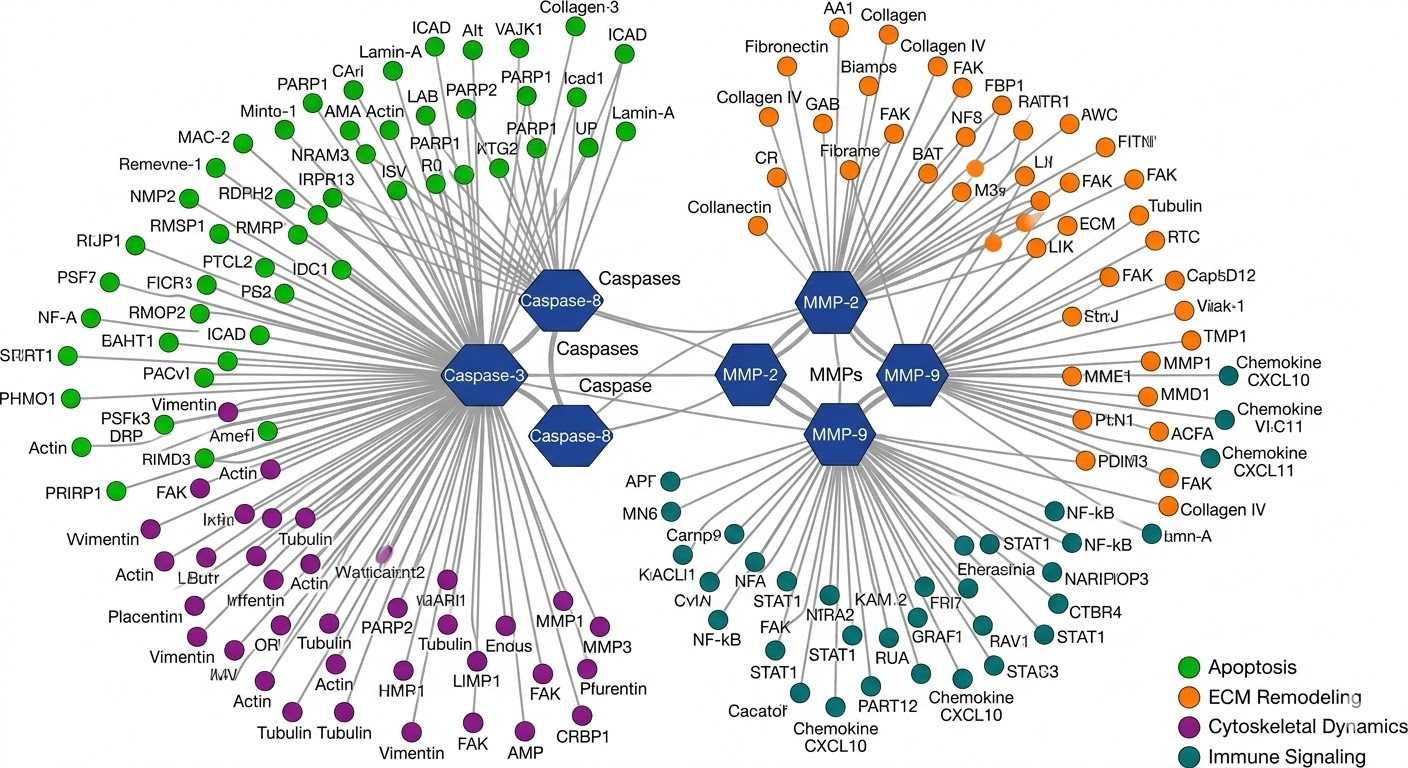
Substrate Pathway Network Graph
Peptide-Level Abundance Comparison
Deliverables
Clients receive comprehensive data packages designed for immediate integration into validation studies and mechanism publications:
- Detailed Cleavage-Site Reports: Exact amino acid positions mapped to the target protein sequences.
- Substrate Candidate Summaries: Ranked lists of endogenous proteins cleaved under your experimental conditions.
- Annotated Peptide Tables: Raw and normalized abundance data for all identified cleavage-derived fragments.
- Comparative Interpretation: Statistical analysis comparing treated versus control proteolysis states.
- Comprehensive Project Report: Full documentation of methods, QC metrics, and analytical parameters.
Frequently Asked Questions
What are the main applications of degradomics in research? +
Degradomics is instrumental in numerous high-impact research areas. It is widely used for biomarker discovery in biofluids, validating drug target inhibitors, mapping host-pathogen interactions during infectious diseases, profiling inflammatory processing signaling, and uncovering the mechanisms behind extracellular matrix remodeling in conditions such as fibrosis and cancer.
What sample types can be used for protease substrate profiling? +
Our degradomics platform supports a wide range of biological and experimental inputs, including cell lysates (treated or untreated), tissue lysates from biopsy or model organisms, biofluids (e.g., serum, plasma, CSF), conditioned media from cell cultures, and in vitro systems using purified or recombinant proteins.
What is degradomics and how does it differ from standard proteomics? +
Standard proteomics uses extensive tryptic digestion to measure the total abundance of intact proteins. Degradomics explicitly targets the non-tryptic, biologically generated fragments and neo-termini produced by endogenous proteases, allowing you to discover specific cleavage sites and active substrates rather than just total protein levels.
What can protease profiling reveal about substrate processing? +
Protease profiling reveals the exact molecular bonds being cut within a biological system. This helps researchers understand whether a protein is being activated, inactivated, or shed from a membrane, providing direct mechanistic insight into enzyme activity that standard expression assays miss.
Can this service identify cleavage sites directly? +
Yes. By isolating and sequencing cleavage-derived peptides (often through N-terminomics enrichment techniques), our high-resolution mass spectrometry platforms can map the exact amino acid position where a protease cleaved the parent sequence.
When is a treated-versus-control design recommended? +
A paired comparative design (e.g., an inhibitor-treated sample versus a vehicle control, or wild-type versus protease-knockout) is highly recommended for biological discovery. It is the most reliable way to filter out background degradation and isolate the specific cleavage events driven by your protease of interest.
Does the workflow support protease-of-interest substrate discovery? +
Yes. We frequently conduct in vitro discovery workflows where complex biological lysates are treated with a purified recombinant protease of interest. Comparing this to an untreated control allows us to rapidly identify the specific physiological substrates targeted by that single enzyme. For researchers exploring broader peptide mapping beyond proteases, our Peptidomics-Based Antigen Discovery and Prediction workflows utilize similar comparative bioinformatic logics.
What outputs are included in the final report? +
The final report includes a comprehensive list of mapped cleavage sites, a ranked table of substrate candidates, comparative abundance heatmaps for treated vs. control samples, and detailed pathway interpretation mapping the affected proteolytic networks.
Case Study: In Vivo N-Terminomic Profiling for Protease Substrate Discovery and Cleavage Mapping
Journal: Frontiers in Molecular Biosciences
Published: 2021
Summary
Using in vivo N-terminomics, researchers profiled the skin degradome of wild-type, ADAMTS2-deficient, ADAMTS14-deficient, and double-deficient mice to identify protease-dependent cleavage events in a physiologically relevant tissue context. By applying N-TAILS-based neo-N-termini enrichment and comparative mass spectrometry, the study moved beyond simple protein abundance profiling and directly captured cleavage-derived peptide evidence linked to protease activity. This approach enabled the identification of extracellular and cell-surface substrate candidates, mapping of cleavage positions in collagen-related proteins, and broader interpretation of protease-regulated biological pathways. The study provides a strong literature-backed example of how degradomics can support substrate discovery, cleavage-site mapping, and mechanism-oriented protease biology research.
Methods
This study used a comparative in vivo degradomics workflow based on N-terminal peptide enrichment.
Researchers applied N-TAILS to mouse skin samples from wild-type animals as well as mice deficient in ADAMTS2, ADAMTS14, or both proteases. By comparing the N-terminomes across these genotypes, they identified neo-N-termini whose abundance patterns were associated with the presence or absence of the target proteases. iTRAQ-based labeling and ratio analysis were used to distinguish cleavage-linked peptide changes from background protein turnover.
The resulting cleavage-derived peptide data were mapped back to parent proteins to define candidate substrates and cleavage regions. For selected intracellular substrate candidates, including actin and vimentin, additional in vitro digestion experiments with purified enzymes were performed to confirm cleavage behavior and strengthen biological interpretation.
Key Technical Features
- Sample types: Mouse skin from wild-type, ADAMTS2-deficient, ADAMTS14-deficient, and double-deficient animals
- Protease context: Comparative in vivo profiling of ADAMTS2- and ADAMTS14-dependent proteolysis
- Enrichment strategy: N-TAILS-based neo-N-termini enrichment for degradome analysis
- Quantitative design: iTRAQ labeling with ratio-based comparison across genotypes
- Core output: Cleavage-derived neo-N-termini, candidate substrates, and mapped cleavage regions
- Biological interpretation: Extracellular substrate discovery and pathway-level analysis of protease-regulated processes
- Orthogonal follow-up: In vitro cleavage confirmation for selected substrate candidates
Creative Proteomics can offer degradomics and protease profiling services that support similar research workflows, including:
- Comparative degradomics for treated-versus-control or perturbation-based study designs
- N-terminomics workflows for neo-N-termini enrichment and cleavage discovery
- Cleavage-site mapping for exact protease processing analysis
- Protease substrate discovery in tissues, cell lysates, conditioned media, or in vitro systems
- Comparative cleavage profiling across biological conditions
- Bioinformatic interpretation linking substrate candidates to pathways and protease activity patterns
These services support biomarker discovery, protease mechanism research, target validation, and broader studies of regulated proteolysis.
Results
Comparative N-Terminomics Revealed a Broad Protease-Dependent Degradome
The study identified a total degradome of 437 proteins associated with ADAMTS2 and/or ADAMTS14 activity, including 68 extracellular and cell-surface proteins that were especially relevant for secreted protease biology. This demonstrates the value of comparative degradomics for moving from complex tissue samples to structured substrate candidate lists.
Extracellular Substrate Candidates and Pathway Signals Were Recovered
Among the identified cleavage-linked proteins, many were associated with collagen organization, extracellular matrix biology, lipoprotein regulation, and inflammatory or immune-related processes. This highlights how degradomics can connect cleavage events to higher-level biological mechanisms rather than reporting peptide changes alone.
Cleavage Site Mapping Clarified Collagen Processing Biology
The N-terminomic data mapped cleavage-associated regions in fibrillar collagen substrates and revealed processing patterns in type I and type V collagens, including both known and additional candidate cleavage sites. These findings illustrate how neo-termini analysis can refine substrate processing models at amino-acid-level resolution.
Orthogonal Follow-Up Strengthened Substrate Interpretation
In addition to extracellular proteins, actin and vimentin were identified as potential substrates. Follow-up in vitro cleavage experiments with purified ADAMTS2 and ADAMTS14 supported these observations, showing how degradomics discoveries can be extended into targeted validation experiments.
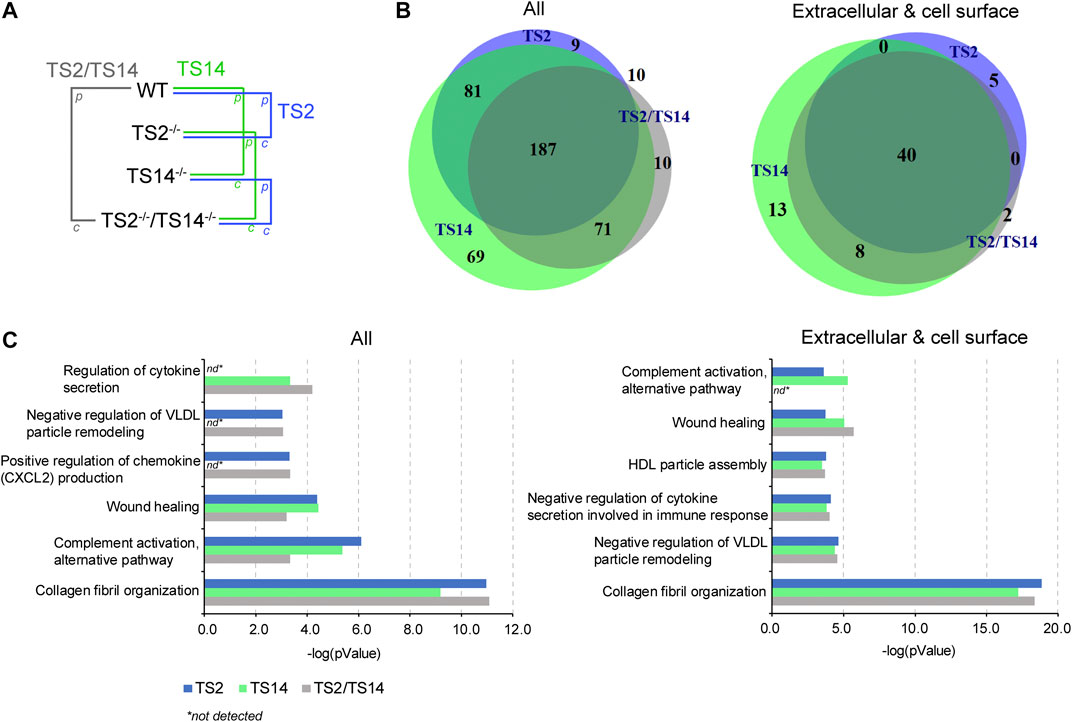
Comparative N-terminomic profiling of wild-type and protease-deficient mouse skin reveals ADAMTS2- and ADAMTS14-dependent substrate candidates and associated biological processes.
Reference
- Leduc, Cédric, et al. "In vivo N-Terminomics Highlights Novel Functions of ADAMTS2 and ADAMTS14 in Skin Collagen Matrix Building." Frontiers in Molecular Biosciences 8 (2021).