Why Potent Peptide Leads Still Fail in Preclinical Development
In therapeutic peptide discovery, developers frequently encounter a frustrating paradox: a peptide lead exhibits exceptional in vitro target affinity and potency, yet completely fails to demonstrate in vivo efficacy.
More often than not, this is a developability failure rather than a pharmacological one. Unlike many small molecules, therapeutic peptides are often cleared through proteolysis, tissue-specific peptidases, renal filtration, and matrix-dependent degradation pathways that are not captured by microsomal CYP-focused assays alone. A candidate might show excellent stability in generic liver microsome screens—giving a false sense of security—but degrade within minutes upon entering systemic circulation due to ubiquitous plasma peptidases. Furthermore, when medicinal chemists introduce sequence modifications to fix the issue, the resulting changes in stability are often unpredictable without knowing exactly where the original sequence failed.
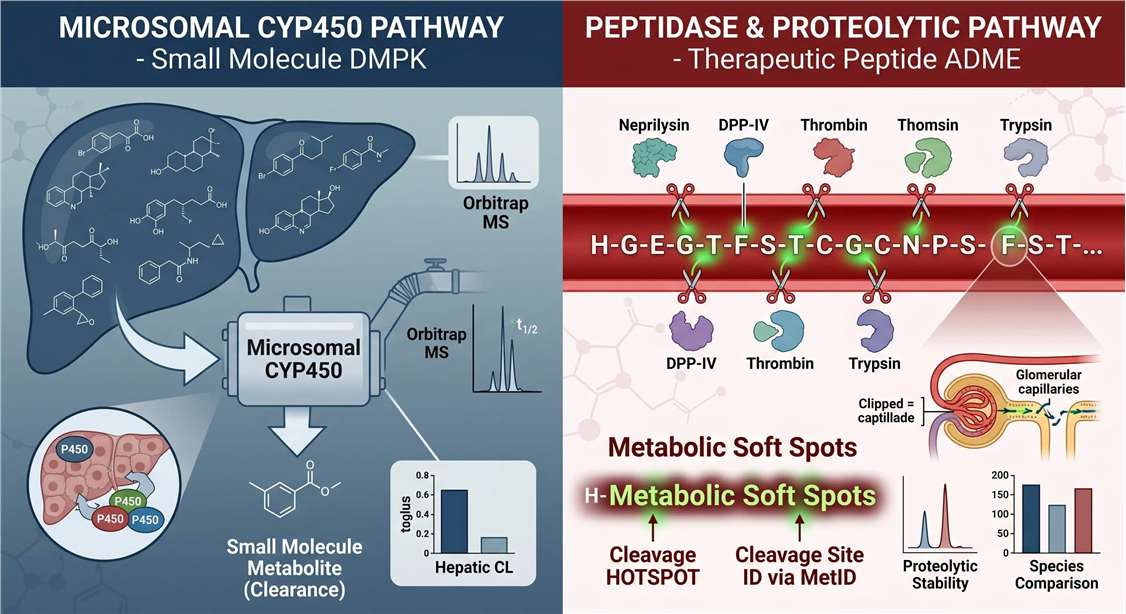
In peptide drug development, failure is usually driven by metabolic vulnerability. Understanding the specific mechanism of this vulnerability is the first critical step toward engineering a successful drug.
Decision Matrix: Why Standard Small Molecule ADME Fails for Peptides
Many drug developers mistakenly route their peptide leads through generic small-molecule DMPK workflows. However, the physicochemical properties and clearance mechanisms of peptides are fundamentally different. Our peptide-specific metabolic stability service is designed around these unique biological realities:
| Parameter | Standard Small Molecule ADME | Peptide-Specific Stability Profiling |
|---|---|---|
| Primary Metabolism Driver | CYP450 Enzymes (Phase I/II) | Endopeptidases & Exopeptidases |
| Most Critical In Vitro Model | Liver Microsomes | Plasma/Serum, S9 Fraction, Lysosomes |
| Analytical Challenge | Simple mass shifts (e.g., +16 Da for oxidation) | Complex fragmentation, multiple cleavage sites, and multiple charge states |
| Primary Deliverable | Clearance rate (CLint) and half-life | Exact cleavage site map (Soft Spot ID) |
| Required Instrumentation | Standard LC-UV or low-res LC-MS | Ultra-high-resolution MS/MS (e.g., Orbitrap) |
Recommended Research Applications
This platform is specifically engineered to bridge the gap between discovery and preclinical development. It is the ideal methodological choice for:
Beyond Half-Life: Find the Metabolic Soft Spots That Limit Your Peptide Lead
Generic ADME services often treat peptides like small molecules, providing a single half-life (t1/2) value and an intrinsic clearance (CLint) rate. While useful for high-throughput screening, a simple "fast-degrading" label does not tell a peptide chemist how to fix the molecule.
This is where our platform diverges from standard stability assays. We focus heavily on Metabolite Identification (MetID) and Metabolic Soft Spot Mapping. By leveraging ultra-high-resolution Orbitrap mass spectrometry, we do not just track the disappearance of the parent peptide; we actively capture and sequence the resulting degradation fragments.
We provide the "why" behind the half-life by delivering:
- Exact Cleavage Sites: Identifying the precise amide bonds targeted by endopeptidases.
- Metabolite Identity: Sequencing the resulting truncated fragments.
- Degradation Routes: Mapping the sequential kinetic pathway of primary and secondary cleavage events.
- Species Differences: Highlighting where human protease activity diverges from preclinical rodent models.
These results are reported as sequence-level cleavage maps, metabolite tables, and time-resolved degradation pathways that directly support analog ranking and redesign decisions.
Which Stability Model Best Answers Your Development Question?
Selecting the right biological matrix is essential. Because peptides do not rely solely on hepatic metabolism, their degradation is highly context-dependent. We offer targeted models to answer your specific developmental questions:
| Development Question | Recommended Model | Why It Matters |
|---|---|---|
| Will my peptide survive systemic circulation? | Plasma / Serum | Assesses blood protease liability |
| Is hepatic metabolism contributing? | S9 / Hepatocytes | Captures broader liver-associated metabolism |
| Is local tissue degradation a risk? | Tissue Homogenates | Reveals organ-specific protease exposure |
| Is oral delivery feasible? | GI Enzyme Panel | Simulates gastric and intestinal degradation |
| Do in vitro findings match animal exposure? | In vivo PK matrix support | Bridges discovery assays and PK interpretation |
From Cleavage Mapping to SAR: How We Turn MetID into Actionable Optimization Insight
Our goal is to serve as a consultative extension of your medicinal chemistry team. We structure our data to directly inform your Structure-Activity Relationship (SAR) campaigns.
When you submit an analog series, we align the MetID data to reveal why certain analogs outperform others. We identify the specific dipeptide bonds acting as cleavage hotspots. For example, if one analog replaces a vulnerable L-amino acid with a D-amino acid and shows delayed cleavage at the original hotspot, the resulting map can help confirm whether the modification protected the intended site or simply shifted degradation to a secondary position.
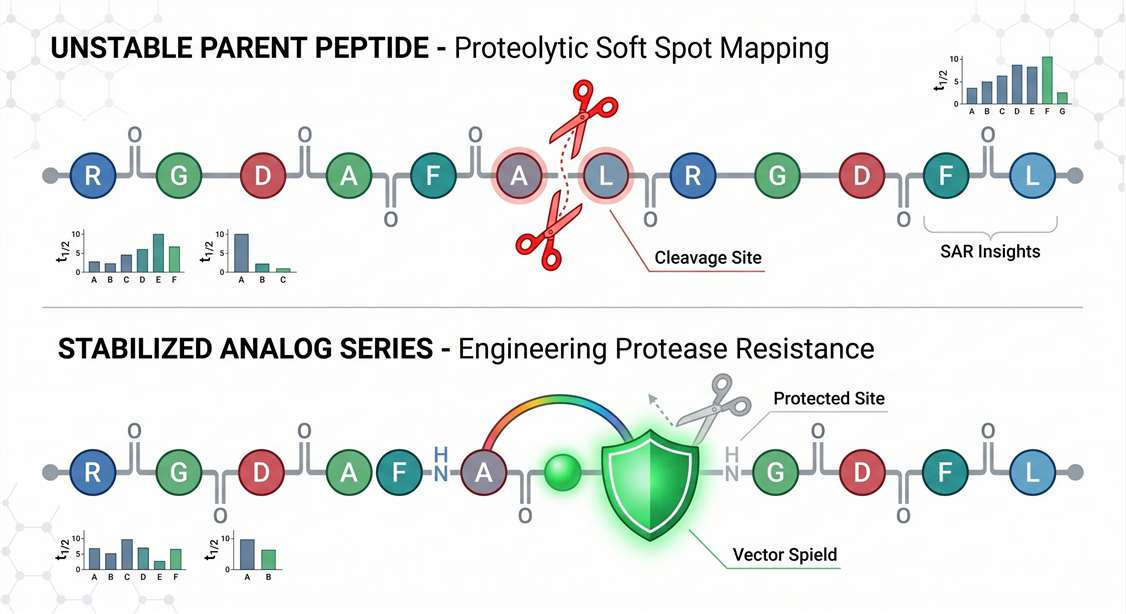
Whether your team relies on custom peptide synthesis to introduce a stapled bridge or a lipid tail, this consultative approach turns raw mass spectrometry data into actionable redesign clues, accelerating your path to a stable lead.
Supported Therapeutic Peptide Formats
Modifying a peptide to improve its stability dramatically alters its ionization, fragmentation behavior, and chromatographic properties. Standard proteomics workflows often fail to detect these engineered molecules. Our mass spectrometry workflows are custom-optimized to support complex therapeutic formats, including:
- Linear Peptides (Endogenous analogs and synthetics)
- Cyclic Peptides (Disulfide-rich, head-to-tail, thioether bonds)
- Stapled Peptides (Hydrocarbon-stapled alpha helices)
- PEGylated Peptides (Varying chain lengths)
- Lipidated Peptides (Cholesterol or fatty-acid conjugated)
- Peptide-Drug Conjugates (PDCs)
- Non-Natural / D-Amino-Acid Peptides
Feasibility and assay optimization may vary depending on peptide hydrophobicity, conjugation chemistry, charge state, and matrix background.
Advanced Hardware & Workflow for Peptide Stability Profiling
To ensure absolute confidence in our cleavage site mapping, our workflow is powered by industry-leading analytical hardware and rigorous quality control standards:
Incubation & Sampling
Physiological alignment
Quenching & Extraction
Matrix interference removal
LC-MS/MS Acquisition
High-res Orbitrap & Triple Quad
Bioinformatics & QC
Soft spot identification
1
Incubation & Sampling
The peptide is spiked into the chosen matrix (e.g., pooled human plasma) at physiological temperatures, with aliquots taken at customized kinetic time points (e.g., 0, 15, 30, 60, 120, 240 mins).
2
Quenching & Extraction
Rigorous sample cleanup using acid quenching and Solid Phase Extraction (SPE) or protein precipitation to halt enzymatic activity and remove matrix interferences.
3
LC-MS/MS Acquisition
Quantitation: Performed on high-sensitivity Triple Quadrupole LC-MS/MS platforms to track parent peptide depletion accurately. Where appropriate, targeted quantitation can be supported using reference standards or internal standards to improve time-course accuracy.
MetID Mapping: Executed on ultra-high-resolution Orbitrap Exploris™ or Eclipse™ systems using HCD/ETD fragmentation to capture comprehensive sequence data of all degradation products.
MetID Mapping: Executed on ultra-high-resolution Orbitrap Exploris™ or Eclipse™ systems using HCD/ETD fragmentation to capture comprehensive sequence data of all degradation products.
4
Bioinformatics & QA/QC
Software-assisted fragment matching is manually reviewed by expert mass spectrometrists. We apply stringent False Discovery Rate (FDR < 1%) controls and retention-time alignment to ensure artifact-free soft spot identification.
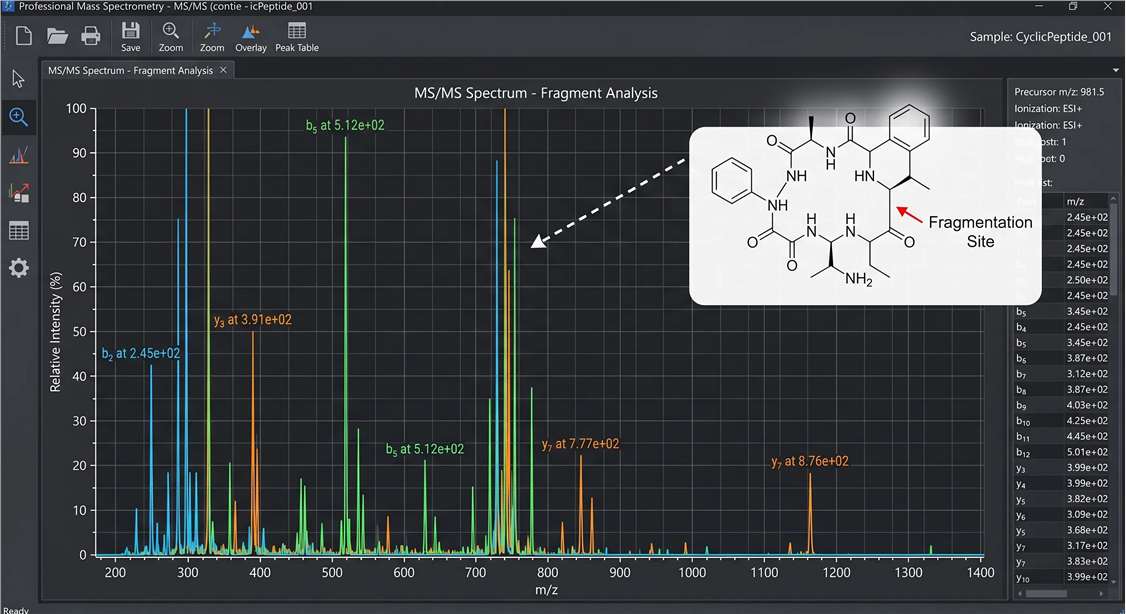
Demo Results: Visualizing Cleavage Dynamics
To ensure the highest biological confidence, we provide multi-dimensional data visualizations that validate structural stability. These are examples of the definitive, actionable insights you receive.
High-Resolution MetID Spectrum
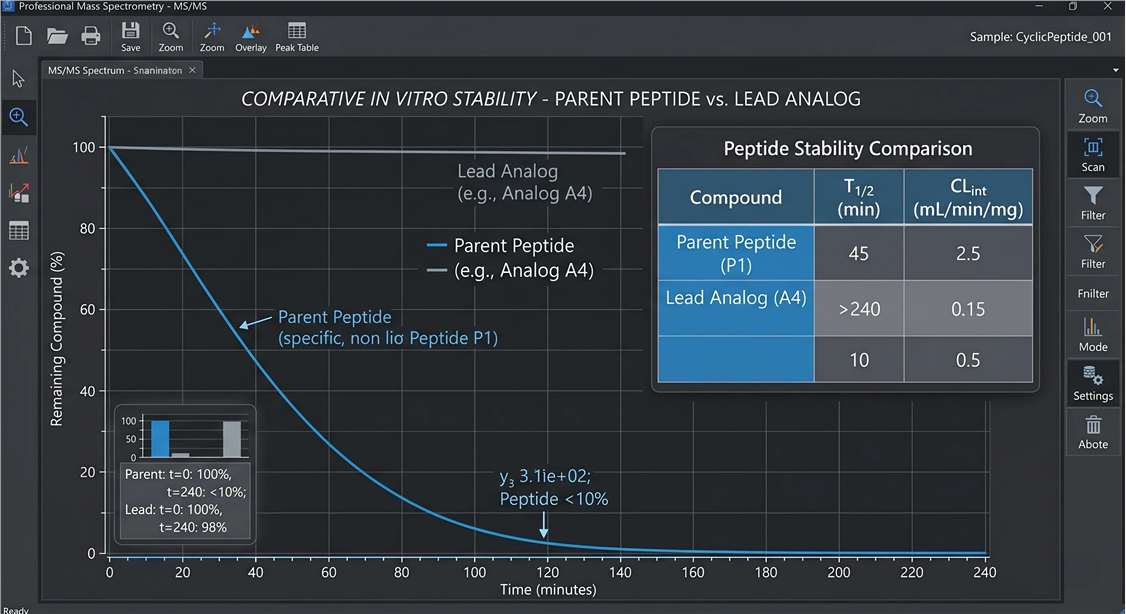
Kinetic Degradation Curves
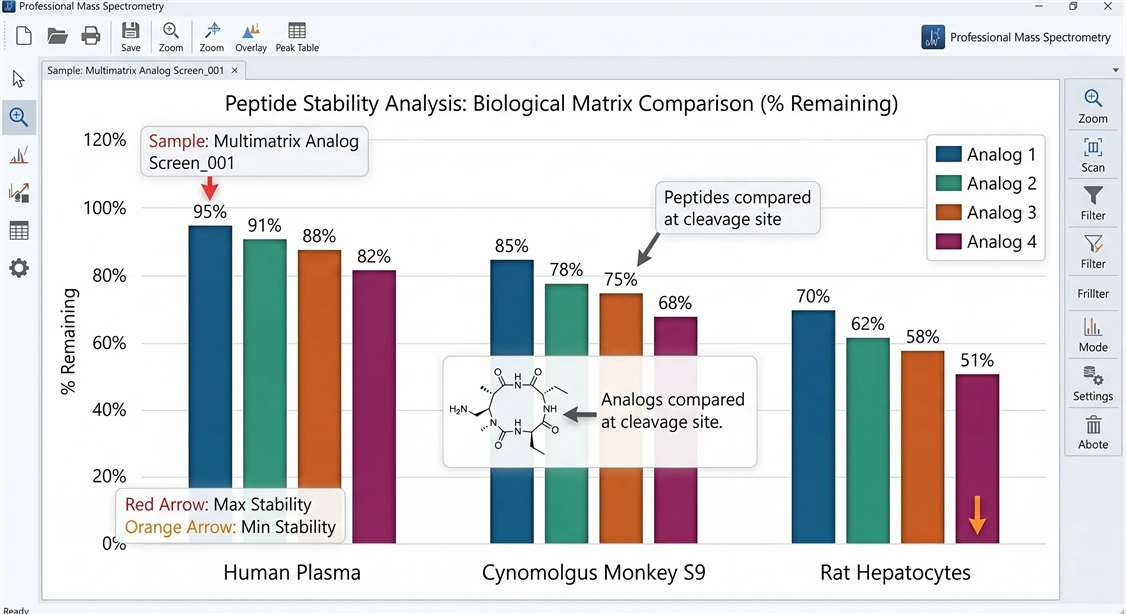
Cross-Species Stability
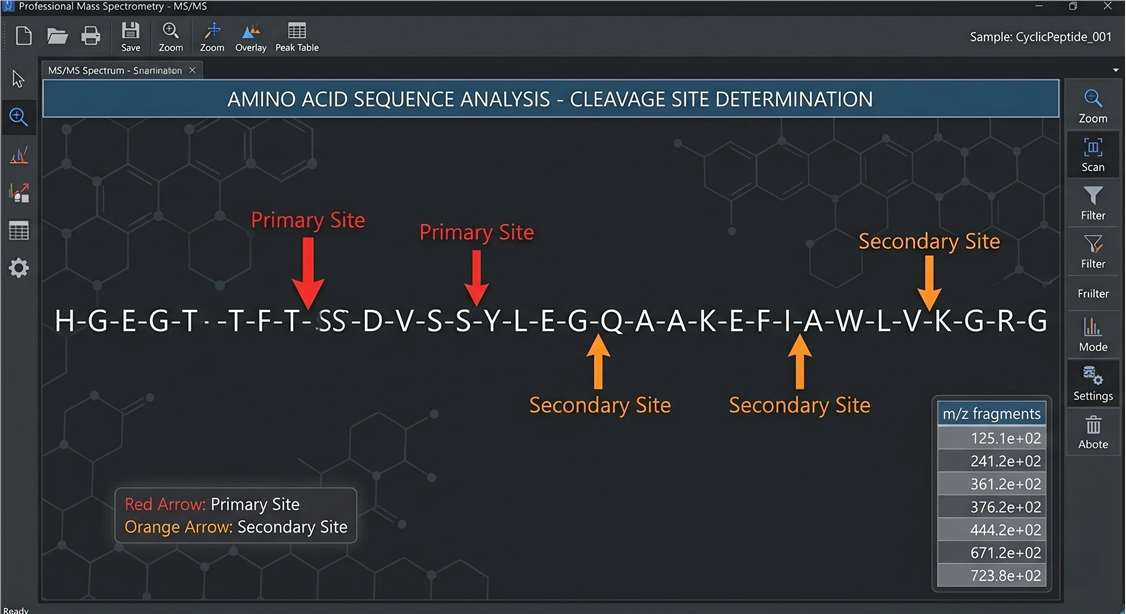
Cleavage Site Map (Soft Spot ID)
Typical Deliverables for Lead Optimization Decisions
We deliver strategic data packages designed to support definitive Go/No-Go decisions. Our typical reporting turnaround time is structured to keep pace with rapid SAR cycles. Your deliverables include:
- Degradation Curves: Kinetic plots tracking the percentage of remaining parent peptide over time.
- t1/2 and CLint Values: Calculated half-life and intrinsic clearance metrics for direct analog ranking.
- Cleavage Site Map: Visual sequence schematics explicitly highlighting the identified primary and secondary proteolytic cleavage sites.
- Metabolite ID Table: A detailed structural table of all identified metabolites, including m/z, retention times, and sequence assignments.
- Cross-Species Comparison: Comparative analytics showing differences in degradation rates and pathways between human, rat, mouse, or NHP models.
- Modification Recommendation Clues: Bioanalytical insights highlighting which specific residues remain vulnerable.
- Matrix / Species Summary Table: Consolidated comparison across tested matrices and species for faster project review.
- Study Summary Report: A concise interpretive report summarizing major liabilities, soft spots, and recommended next-step considerations.
- Raw Data Access: Vendor-native raw files and processed data tables available upon request.
Sample Requirements and Study Design Recommendations
Optimal assay performance requires proper planning and high-quality starting material.
| Material | Minimum Amount | Required Information | Format / Purity | Shipping |
|---|---|---|---|---|
| Synthetic Peptide | 1.0 – 5.0 mg | Sequence, MW, modification map | Lyophilized or stock; high purity preferred | Ambient / Dry ice |
| Analog Series | Case by case | Analog list and intended comparison | Prefer batch submission | Dry ice |
| In Vivo Matrices | 100 μL+ | Species, matrix type, collection conditions | Plasma, Serum, Urine, Bile | Dry Ice |
Study Design Recommendations:
- Sequence Annotation: Clients must provide the exact amino acid sequence. For modified peptides, please specify the exact location and chemistry of stapling, PEGylation, lipidation, cyclization, or non-canonical residues to support accurate mass assignment and metabolite interpretation.
- Pilot Feasibility: For highly modified or unusually hydrophobic peptides (like lipidated species), we recommend a brief pilot feasibility run to optimize LC-MS extraction and ionization conditions before executing a multi-time-point kinetic study.
- Analog Screening Design: When comparing multiple analogs, submitting them as a batch ensures they are run under identical LC-MS/MS conditions and matrix lots, significantly reducing batch-to-batch variation and strengthening comparative confidence.
Disclaimer: All services and analytical platforms described are intended for Research Use Only (RUO). Not for use in diagnostic procedures.
Can you pinpoint exact cleavage sites in therapeutic peptides? +
Yes. By utilizing high-resolution Orbitrap mass spectrometry, we sequence the degradation products formed during the stability assay. By comparing these fragment sequences to your parent peptide, we can map the exact amide bonds that are being targeted by endogenous proteases.
Which model is more informative for peptides: plasma, S9, hepatocytes, or microsomes? +
For most systemically administered peptides, plasma/serum is the most informative primary model, as blood peptidases are the first and most aggressive barrier to exposure. S9 fractions and hepatocytes are excellent secondary models for capturing liver-specific peptidase activity. Microsomes are generally the least informative for peptides, as they are optimized for CYP450 small-molecule metabolism rather than proteolysis.
Do you support cyclic, stapled, PEGylated, or D-amino-acid-containing peptides? +
Absolutely. We specialize in non-canonical and highly modified therapeutic formats. Our bioanalytical team custom-tunes the LC gradient, ionization parameters, and extraction methods to accommodate the unique physicochemical properties of lipidated, stapled, and cyclic peptides.
Can you compare metabolic stability across human, rodent, and NHP matrices? +
Yes. We routinely run parallel stability assays using matched matrices (e.g., plasma or S9) from Humans, Mice, Rats, and Non-Human Primates (Cynomolgus/Rhesus). This is highly recommended to identify species-specific degradation pathways before initiating expensive in vivo animal studies.
Can the results guide next-round peptide optimization? +
Yes. Providing actionable data is the core purpose of this service. By identifying exactly which residues or bonds are vulnerable to cleavage, your medicinal chemistry team can strategically introduce specific modifications—such as N-methylation, steric shielding, or stereochemical inversion—precisely where they are needed to engineer a more stable candidate.