The Proteogenomic Advantage: Bridging Genomics and Immunopeptidomics
The Biological Bottleneck of In Silico Prediction
While next-generation sequencing (NGS) rapidly catalogs tumor-specific somatic mutations, standard algorithmic prediction tools (e.g., NetMHCpan) primarily assess theoretical HLA binding affinity. They cannot accurately predict complex upstream biological processing—such as proteasomal cleavage, TAP transport, and ERAP trimming. Relying solely on these algorithms ignores this biological bottleneck, often resulting in false-positive rates exceeding 90% and wasting resources on targets that are never physically presented on the cell surface.
Overcoming Standard Database Constraints
Standard HLA Peptidomics Analysis relies on searching mass spectrometry data against wild-type reference proteomes (like UniProt), which inherently lack patient-specific somatic mutations. Proteogenomics bridges this analytical gap. By translating a tumor's specific WES and RNA-seq data into a customized variant FASTA database, our high-resolution mass spectrometers can directly identify the physical presence of mutated, tumor-restricted neoantigens.
Key Application Areas in Immuno-Oncology
Our multi-omics neoantigen discovery platform is tailored for translational oncology teams developing precision immunotherapies:

Personalized Cancer Vaccines
Formulate highly specific vaccines based strictly on MS-validated, physically presented neoepitopes to maximize robust CD8+ and CD4+ T cell activation and bypass immune tolerance.
TCR-T Cell Therapy Target Discovery
Prevent on-target, off-tumor toxicities by acquiring definitive physical proof of tumor-restricted presentation, ensuring safely engineered, highly specific T-cell receptors.
Immune Checkpoint Inhibitor (ICI) Biomarkers
Profiling matched pre- and post-treatment tumor biopsies reveals the true presented neoantigen burden and identifies potential mechanisms of acquired resistance to PD-1/PD-L1 blockade therapies.
Comprehensive Proteogenomic Discovery Solutions
We offer a modular, end-to-end multi-omics framework. Whether you need us to handle both the genomic sequencing and the mass spectrometry from a raw tissue biopsy, or you wish to provide your own internally generated NGS data for us to integrate, our computational pipeline adapts seamlessly to your specific operational requirements.
Detectable Neoantigen and Cryptic Peptide Types
A key advantage of integrating deep RNA-seq with high-resolution mass spectrometry is the unparalleled ability to discover non-canonical and highly immunogenic targets that DNA sequencing alone fundamentally cannot detect. Because these non-canonical peptides bypass central thymic tolerance, they often harbor the highest immunogenic potential.
| Mutation / Antigen Type | Biological Origin | Proteogenomic Detection Strategy |
|---|---|---|
| Single Amino Acid Variants (SAAVs) | Point mutations (SNVs) occurring within exonic, protein-coding regions. | Direct sequence mapping of mass spectra to WES-derived variant databases. |
| Frameshift Mutations | INDELs causing a shift in the standard ribosomal reading frame. | Translation of novel, often highly divergent downstream peptide sequences. |
| Gene Fusions | Chromosomal translocations leading to chimeric transcripts. | Detection of novel chimeric peptides physically spanning the fusion junction. |
| Alternative Splicing Variants | Splice site mutations or epigenetically dysregulated splicing. | RNA-seq guided detection of exon-skipping or intron-retention events. |
| Cryptic Peptides | Non-coding regions (ncRNA, 5'/3' UTRs) becoming aberrantly translated. | Searching MS spectra against comprehensive 6-frame translated RNA-seq data. |
Multi-Omics Pipeline for Mutated Peptide Identification
Our end-to-end workflow is subject to stringent quality control checkpoints at both the genomic and proteomic levels.
Multi-Omics Sample Processing
NGS Acquisition & Transcript Validation
Variant Database Construction
Ultra-Sensitive LC-MS/MS
Proteogenomic Alignment & Bioinformatics
1
Multi-Omics Sample Processing
DNA, RNA, and HLA complexes are extracted from the supplied tumor tissue (and matched normal tissue). High-quality NGS libraries and highly purified HLA ligand eluates are prepared in parallel.
2
NGS Acquisition & Transcript Validation
WES identifies somatic variants, while RNA-seq confirms that the mutated gene is actively transcribed and expressed in the tumor microenvironment.
3
Variant Database Construction
A highly specific FASTA database is compiled, containing both the wild-type human proteome and the sample-specific mutated sequences. Decoy sequences are specifically generated to account for the unspecific enzymatic cleavage typical of HLA ligands.
4
Ultra-Sensitive LC-MS/MS
The eluted HLA peptides are analyzed using Data-Dependent Acquisition (DDA) or Data-Independent Acquisition (DIA) on advanced high-resolution mass spectrometers.
5
Proteogenomic Alignment & Bioinformatics
MS/MS spectra are searched against the custom database. The bioinformatics pipeline evaluates mass accuracy, fragmentation patterns, and retention times to unambiguously identify mutated peptides, filtering out wild-type equivalents.
Advanced Bioinformatics & Variant Prioritization
Detecting a true somatic mutation in mass spectrometry data requires far greater statistical and computational rigor than standard global proteomics. Because neoantigens are often expressed at extremely low copy numbers per cell, our bioinformatics unit employs advanced algorithms specifically designed to separate true immunopeptidomic signal from inherent background noise.
Sample Requirements for Multi-Omics Integration
Proteogenomic integration requires sufficient biological material to simultaneously extract DNA, RNA, and HLA-bound peptides without compromising the yield of any single modality. The following guidelines ensure sufficient input for high-confidence multi-omics profiling.
| Sample Type | Genomics Input (WES/RNA-seq) | Proteomics Input (HLA-MS) | Preservation Notes |
|---|---|---|---|
| Fresh Frozen Tumor | ≥ 30 mg | ≥ 50 – 100 mg | Snap-frozen; essential for integrated multi-omics. |
| Matched Normal Tissue | ≥ 30 mg | N/A | Highly recommended for somatic variant calling filtering. |
| Matched PBMC / Blood | ≥ 2 mL (Whole Blood) | N/A | Alternative to normal tissue for establishing the baseline germline genome. |
| Pre-Extracted Data | ≥ 1 µg DNA / ≥ 2 µg RNA | N/A | DNA/RNA must meet strict quality standards (e.g., RIN >7.0). FastQ or BAM files are accepted. |
Note: Formalin-Fixed Paraffin-Embedded (FFPE) tissues are strictly incompatible with HLA ligandome immunoaffinity purification.
Demo Results: High-Confidence Proteogenomic Evidence
Our data packages deliver clear, publication-quality visualizations that bridge the genomic data with the mass spectrometry physical evidence, ensuring your downstream decisions are backed by transparent data.
Genome-to-Proteome Mapping (IGV Tracks)
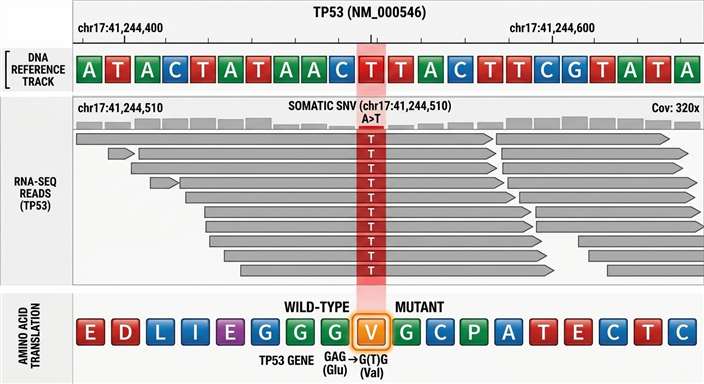
Visually confirms that the physical peptide detected by mass spectrometry perfectly aligns with the somatic mutation called and expressed in the transcriptomic data, bridging the central dogma from DNA to presented protein.
Annotated MS2 Spectrum of Mutated Peptides
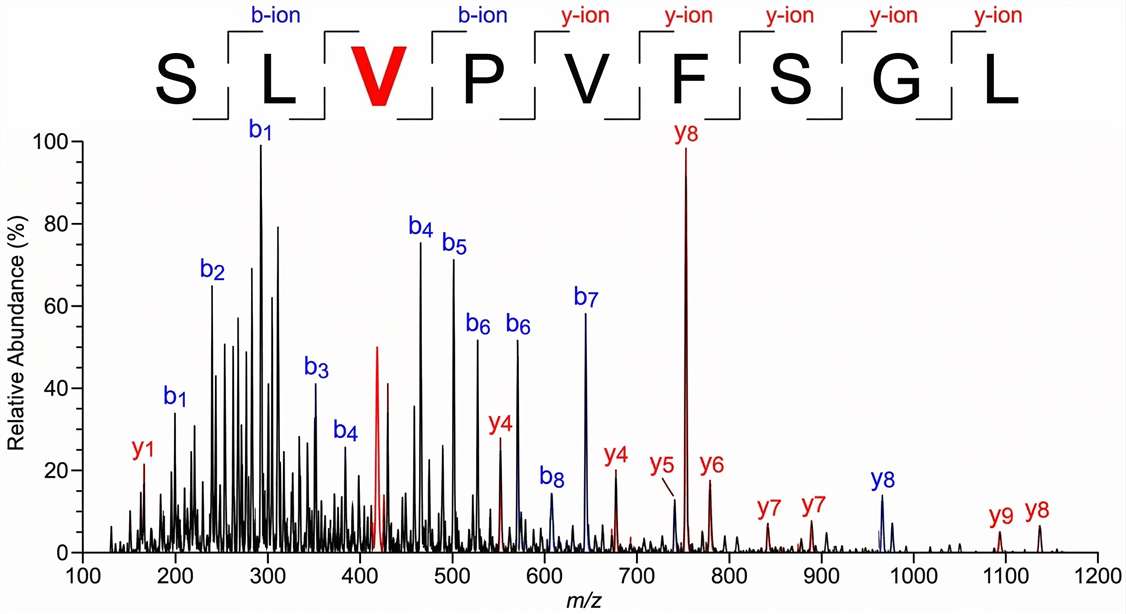
Pinpoints the exact mass shift caused by the mutation in the continuous b-ion and y-ion series, providing unambiguous proof of the peptide's exact amino acid sequence and the precise position of the somatic mutation.
Prediction vs. MS Validation Overlap

Highlights the critical filtering power of mass spectrometry via comparative Venn diagrams, demonstrating the vast number of genomic predictions that biologically fail to be physically presented on the HLA complex.
Binding Affinity Distribution
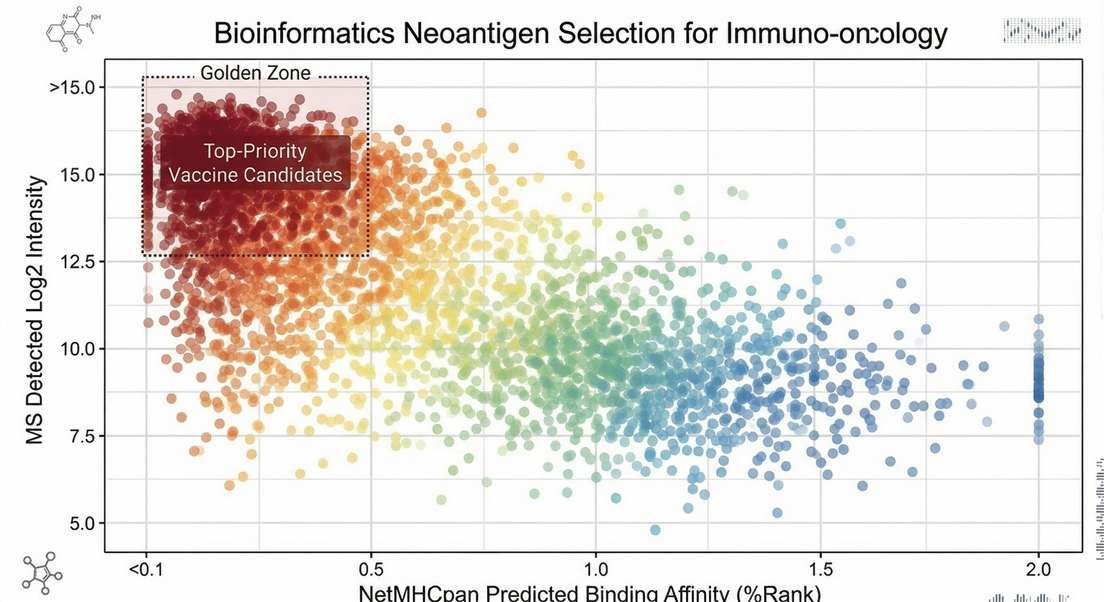
Helps prioritize clinical candidates by correlating MS-detected peptide intensities (abundance) with NetMHCpan predicted binding affinities, identifying neoantigens that possess both strong physical presentation and high biological stability.
Service Level Selection: Proteogenomics vs. Targeted Validation
Depending on your internal bioinformatics capabilities and your current pre-clinical project stage, we offer distinct service architectures to match your needs.
| Feature | Proteogenomics-Driven Discovery | Targeted Neoantigen Validation |
|---|---|---|
| Primary Objective | Unbiased, global discovery of novel mutated targets directly from patient tissue. | Exact physical verification of a predefined list of AI-predicted candidates. |
| Required Sample Inputs | Tissue for MS + Matched Tissue/Data for NGS (WES/RNA-seq). | Tissue for MS + Pre-selected Candidate Peptide List. |
| Mass Spectrometry Platform | Orbitrap Astral™ / timsTOF Pro (Maximized scanning depth). | Orbitrap Exploris™ 480 / Q Exactive HF-X. |
| Acquisition Mode | Untargeted (Data-Dependent DDA / Data-Independent DIA). | Targeted (Parallel Reaction Monitoring - PRM / SureQuant™). |
| Database Strategy | We build and search against a custom variant FASTA from your multi-omics data. | No FASTA search required; we synthesize heavy-isotope internal standards. |
| Sensitivity Focus | Broad proteome coverage to identify dominant naturally presented mutations. | Absolute maximum sensitivity for ultra-low abundance specific targets. |
| FDR Control Strategy | Strict <1% PSM-level FDR using non-tryptic decoy databases. | Direct retention time and fragmentation matching against synthetic standards. |
| Best Suited For | Early-stage target discovery, exploratory research, and neoantigen burden profiling. | Late-stage pre-clinical validation, IND-enabling data, and final vaccine formulation. |
Deliverables
Your final multi-omics data package includes comprehensive reporting tailored specifically for downstream immunological assessment and IND-enabling documentation.

- Mutated Peptide Evidence List: CSV files detailing identified neoantigens, sequences, source genes, mutation types, and mass spectrometry confidence scores.
- Variant Annotation Sheet: Deep integration of genomic loci, transcript expression levels (TPM), and peptide-spectrum match (PSM) validation data.
- HLA Binding Prediction Summary: Detected peptides mapped against the patient's derived HLA haplotype with calculated binding affinities.
- Raw Multi-Omics Data: Full access to raw FASTQ/BAM files (if sequenced by us) and high-resolution mass spectrometry RAW files.
- Executive Summary PDF: Graphical highlights including length distributions, mutation proportions, sequence motifs, and comparative Venn diagrams.
Frequently Asked Questions (FAQ)
Why do I need to provide both genomic (WES/RNA-seq) and tissue samples for this service? +
Proteogenomics inherently requires both modalities to function. The genomic data (DNA/RNA) is used to locate somatic mutations and build the custom search database. The physical tissue sample is used to extract the HLA molecules and perform mass spectrometry to prove which of those mutated sequences are physically presented. Without the genomic data, the mass spectrometer would not know what mutant sequences to look for in the vast background of normal peptides.
Can you use our existing WES/RNA-seq data to build the custom database? +
Yes. If your internal bioinformatics team has already performed WES and RNA-seq, you can securely transfer the raw FASTQ or aligned BAM/VCF files to us. As long as the sequencing meets our depth and quality thresholds, we can utilize your existing data to generate the customized FASTA database, significantly saving you time and redundant sequencing costs.
How do you control the False Discovery Rate (FDR) for mutated peptides, considering they are not in standard human reference databases? +
We apply rigorous statistical controls. When generating the custom FASTA database, our algorithms create corresponding "decoy" sequences specifically tailored to mimic the non-tryptic, unspecific enzymatic cleavage patterns of naturally processed HLA ligands. We enforce a strict <1% FDR at the Peptide-Spectrum Match (PSM) level, ensuring absolute confidence in every reported neoantigen.
What if I only have tumor tissue but no matched normal tissue (e.g., PBMC/healthy tissue)? +
Matched normal tissue is highly recommended because it allows us to precisely filter out patient-specific germline variations, leaving only true tumor-specific somatic mutations. If matched normal tissue is completely unavailable, we can perform tumor-only sequencing and use population databases (such as dbSNP or gnomAD) to filter known germline variants. However, this approach carries a slightly higher risk of reporting private germline variants as neoantigens.
Can this proteogenomic pipeline identify neoantigens derived from gene fusions or alternative splicing, or just SNVs? +
Our pipeline identifies a highly diverse spectrum of mutations. Because we actively integrate RNA-seq data, we are not limited to Single Amino Acid Variants (SAAVs). We can accurately translate and detect highly immunogenic neoantigens resulting from frameshift mutations, gene translocations (fusions), and retained introns or alternative splicing events that are actively transcribed in the tumor microenvironment.
Do you provide HLA binding affinity predictions (e.g., NetMHCpan) alongside the mass spectrometry detection data? +
Yes. Our final bioinformatics deliverable is a functional prioritization matrix. We provide the physical mass spectrometry detection evidence and align it with the patient's HLA typing and algorithmic binding predictions (e.g., NetMHCpan %Rank). This provides a comprehensive view of both physical surface presentation and inherent immunological relevance.
How does this Proteogenomic discovery service differ from standard HLA Peptidomics? +
Standard HLA Peptidomics Analysis maps mass spectrometry data exclusively against wild-type reference databases (like UniProt) to profile the baseline self-antigen landscape. Proteogenomics is a highly specialized, computationally intensive pipeline that requires genomic integration to create bespoke databases, allowing us to actively hunt for tumor-specific somatic mutations that simply do not exist in standard public databases.