Experimental Neoantigen Validation: Overcoming In Silico Prediction Limits
Next-generation sequencing (NGS) tools efficiently catalog the somatic mutational landscape of a tumor. However, translating a raw genomic mutation into a viable, highly immunogenic clinical target presents a formidable biological challenge. Standard in silico prediction algorithms primarily assess whether a mutated peptide sequence possesses the theoretical binding affinity to anchor into a specific HLA allotype groove.
Unfortunately, these algorithms cannot account for the complex upstream intracellular antigen processing machinery—including proteasomal cleavage efficiency, cytosolic degradation, TAP complex transport, and ERAP trimming. Relying solely on algorithmic predictions ignores this fundamental biological bottleneck, resulting in false-positive rates that routinely exceed 90%. To prevent funneling valuable pre-clinical resources into targets that never reach the tumor cell surface, physical tumor neoantigen discovery and validation via mass spectrometry is mandatory.
While standard HLA Peptidomics Analysis profiles the baseline wild-type immune presentation using reference proteomes, our validation service utilizes custom variant databases to provide definitive evidence that a predicted neoantigen is actively processed and naturally presented.
Key Application Areas in Immuno-Oncology
Our integrated discovery and validation platform is specifically engineered for translational oncology teams and biotech innovators prioritizing tumor-specific antigens:
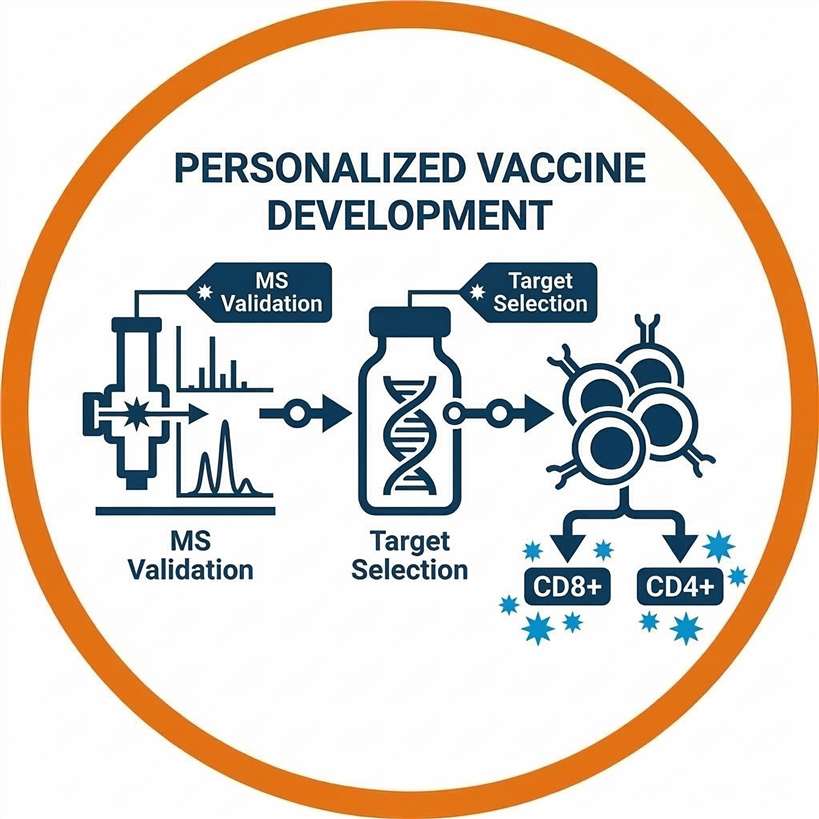
Personalized Cancer Vaccine Development
Developing potent mRNA, DNA, or peptide-based vaccines requires absolute certainty in target selection. Validating that a neoepitope is physically presented ensures that the formulated vaccine elicits a robust, tumor-directed CD8+ and CD4+ T cell response while bypassing central tolerance.
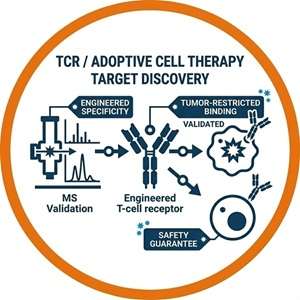
TCR / Adoptive Cell Therapy Target Discovery
T-cell receptor engineered therapies demand targets that are exclusively presented on tumor tissue. Physical MS validation provides the definitive proof of tumor-restricted presentation required to safely engineer TCRs and decisively mitigate the risk of catastrophic on-target, off-tumor toxicities.
Translational Immuno-Oncology Studies
Profiling the physically validated neoantigen burden (as opposed to mere genomic TMB) provides deeper insights into immune escape mechanisms. Comparing pre- and post-treatment tumor biopsies helps identify shifts in antigen presentation and mechanisms of resistance to immune checkpoint inhibitors.
Integrated Discovery and Validation Capabilities
We offer a flexible, multi-layered service architecture designed to accommodate your specific data availability and clinical development stage.
Detectable Tumor-Specific Antigen Types
Integrating RNA-seq with high-resolution mass spectrometry unlocks the ability to discover non-canonical targets that traditional DNA-centric predictions fundamentally overlook.
| Antigen Type | Biological Origin | Validation Strategy |
|---|---|---|
| SNV-Derived Neoantigens | Point mutations in protein-coding exons. | Direct mapping of MS/MS spectra to WES-derived variant databases. |
| Indel-Derived Neoantigens | Frameshift mutations altering the ribosomal reading frame. | Identification of novel downstream peptide sequences via custom FASTA search. |
| Fusion-Derived Neoantigens | Chromosomal translocations producing chimeric transcripts. | Detection of novel peptides physically spanning the junction of fused genes. |
| Splice-Derived Neoantigens | Dysregulated splicing or splice site mutations. | RNA-seq guided mapping of intron-retention or exon-skipping events. |
| Cryptic / Non-Canonical Peptides | Translation of non-coding regions (UTRs, ncRNAs). | Searching MS spectra against comprehensive 6-frame translated RNA-seq data. |
Multi-Tiered Neoantigen Validation Workflow
Our end-to-end workflow rigorously integrates genomic, transcriptomic, and proteomic data streams to seamlessly transition from initial discovery to definitive candidate validation.
Study Design & Input Review
Sequencing & Customized Database
Candidate Discovery & HLA Enrichment
Evidence Integration
Ranking & Reporting
1
Study Design & Input Review
We consult with your team to assess available inputs (tissue, FastQ files, or pre-predicted lists) and determine the optimal validation tier.
2
Sequencing & Customized Database Generation
WES and RNA-seq data are processed to call somatic variants, which are then compiled into a highly specific mutant FASTA database.
3
Candidate Discovery & HLA Enrichment
Intact HLA-peptide complexes are isolated from the tumor tissue lysate, and the natively bound ligands are eluted for ultra-sensitive LC-MS/MS acquisition.
4
Validation-Oriented Evidence Integration
Mass spectra are matched against the custom database. Through our advanced Peptidomics-Based Antigen Discovery and Prediction algorithms, physical detection data is synthesized with HLA binding predictions.
5
Ranking & Reporting
Validated neoantigens are strictly ranked based on MS abundance, binding affinity (%Rank), and transcriptomic expression, delivering a prioritized shortlist for functional follow-up.
Advanced Bioinformatics & Evidence Integration
Validating a neoantigen via mass spectrometry requires computational rigor far beyond standard global proteomics, as mutated peptides are often low-abundance and lack predictable tryptic cleavage sites.
Validation Tiers: Choosing the Right Evidence Level
We stratify our services into distinct validation tiers, allowing you to select the precise level of evidence required for your current research stage.
| Service Tier | Data Inputs Required | Output Evidence Level | Best Suited For |
|---|---|---|---|
| Level 1: Prediction Only | WES / DNA Sequencing | Low: Theoretical HLA binding affinity scores based on genomic variants. | Early exploratory screening; establishing baseline tumor mutational burden. |
| Level 2: RNA-Supported Prioritization | WES + RNA-seq | Medium: Filters predictions to include only actively transcribed mutations. | Refining in silico lists prior to committing to wet-lab validation. |
| Level 3: Proteogenomics Discovery | WES + RNA-seq + Tissue | High: Untargeted MS/MS profiling to discover and validate presented neoantigens globally. | Comprehensive novel target discovery directly from clinical biopsies. |
| Level 4: Targeted Validation | Candidate List + Tissue | Highest: Targeted PRM/SureQuant MS using synthesized heavy-isotope standards for absolute confirmation. | Late-stage pre-clinical validation; finalizing targets for IND-enabling studies. |
Sample and Data Input Requirements
Successful multi-omics evidence integration depends on receiving high-quality tissue or sequencing data.
| Sample / Data Type | Minimum Input | Accepted Formats | Purpose in Pipeline |
|---|---|---|---|
| Tumor Tissue | ≥ 50 mg | Fresh Frozen (No FFPE) | Co-extraction of DNA/RNA and intact HLA complexes. |
| Matched Normal Tissue | ≥ 30 mg | Fresh Frozen or Blood | Filtering out patient-specific germline variations to isolate somatic mutations. |
| Raw Sequencing Data | N/A | FastQ, BAM, or VCF | Utilized if your team has already performed WES and RNA-seq. |
| Predicted Candidate List | N/A | CSV or Excel format | Required only for Level 4 targeted MS-assisted validation workflows. |
Demo Results: Multi-Layered Validation Evidence
Our deliverable packages include publication-ready, Nature-grade data visualizations that clearly demonstrate the rigorous intersection of transcriptomic analysis, predictive algorithms, and physical mass spectrometry validation.
Multi-Omics Target Prioritization Matrix
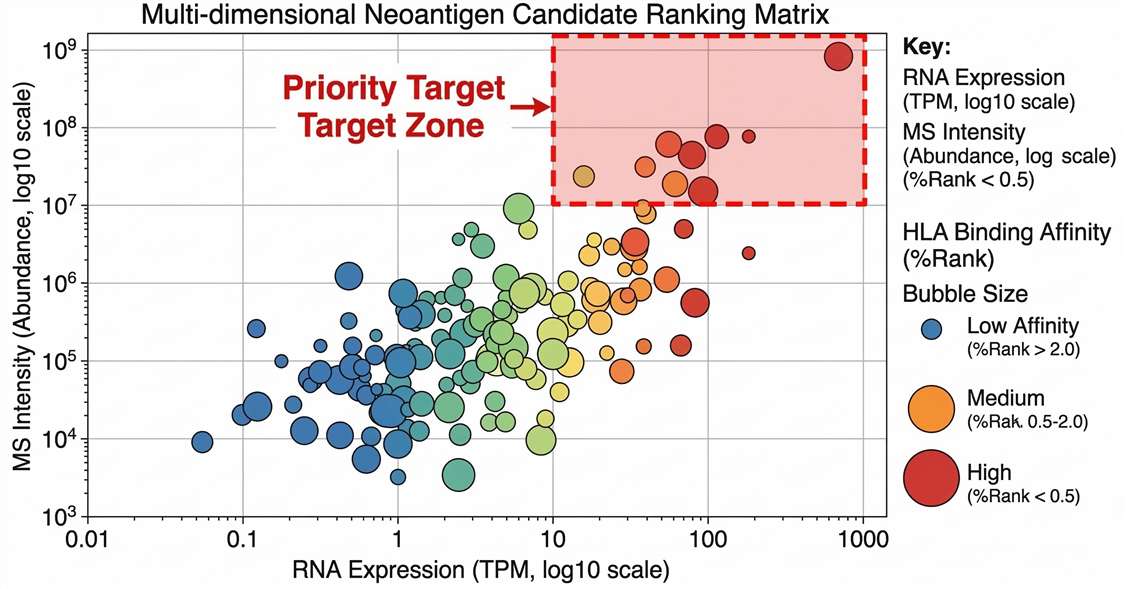
Comprehensive target prioritization: Correlation of transcriptomic expression (log2 TPM) with physical peptide abundance. Marker color indicates HLA-binding stability, pinpointing elite candidates.
False-Positive Filtration Dynamics
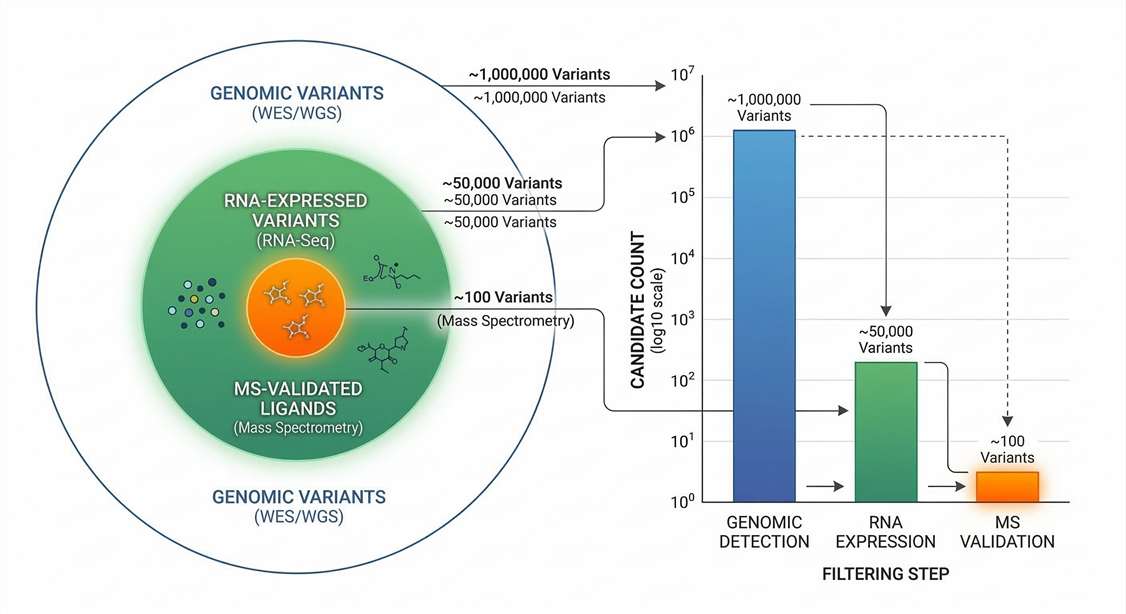
Proteogenomic filtration pipeline: Illustrating the elimination of >90% of in silico predicted candidates through stringent RNA-level and physical MS/MS-level validation.
High-Resolution MS/MS Validation Evidence
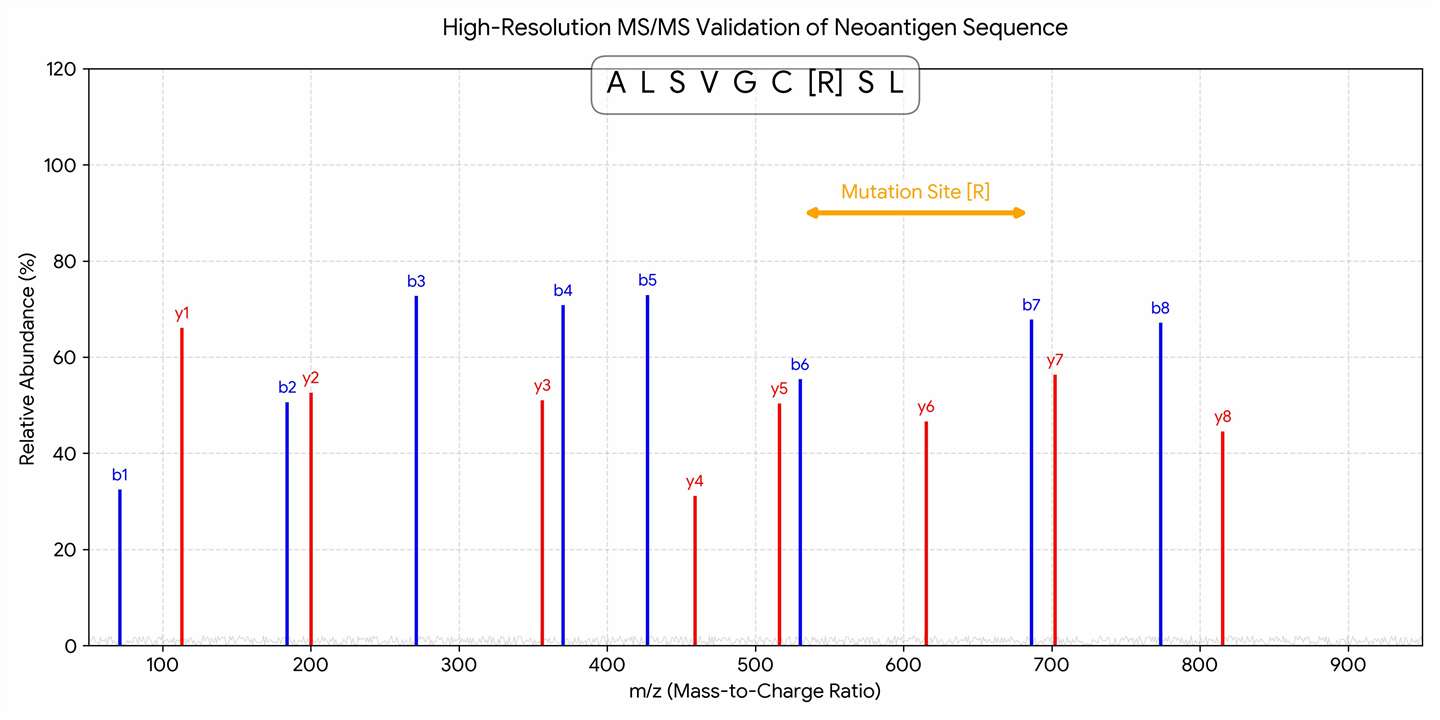
Definitive physical proof: Annotated tandem mass spectrum (MS2) confirming the exact amino acid sequence and position of the tumor-specific somatic mutation.
Genomic Landscape of Validated Ligands
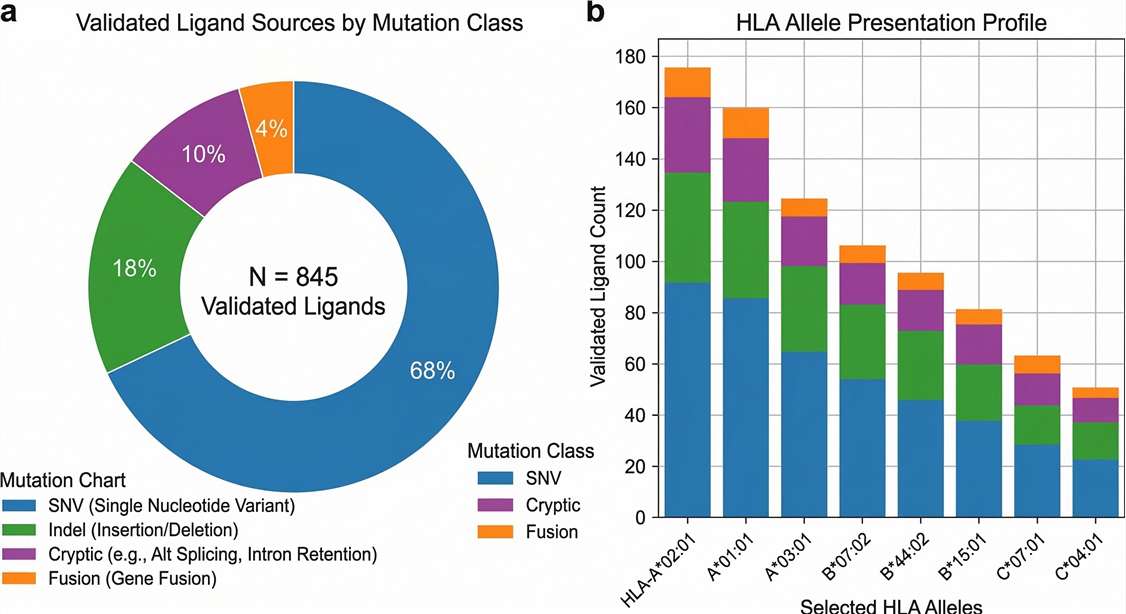
Mutational diversity: Distribution of physically validated HLA ligands, highlighting the successful capture of highly immunogenic non-canonical and structural variants.
Deliverables | What You Will Receive
Your data package is rigorously formatted to facilitate immediate decision-making and seamless transition into downstream functional validation assays.
- Ranked Tumor Neoantigen Candidate Table: A prioritized matrix of validated targets, scored by physical MS abundance, RNA support, and binding affinity.
- Mutation Annotation Sheet: Detailed documentation of the genomic loci, variant type, and surrounding sequence context for each validated peptide.
- HLA Binding Prediction Summary: Computed binding metrics (e.g., NetMHCpan %Rank) mapped against the patient's specific HLA class I/II alleles.
- Peptide-Spectrum Evidence File: High-resolution annotated MS/MS spectra proving the physical sequence identification of top candidates.
- Executive Summary PDF: Comprehensive graphical reports including predicted vs. observed overlap diagrams, mutation class distributions, and methodological quality control metrics.
Case Study: Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry
Journal: Nature Communications
Published: Volume 7, 2016
Summary
Using a multi-omics proteogenomic approach, researchers integrated whole-exome sequencing (WES), transcriptomics, and high-resolution mass spectrometry to identify naturally presented tumor-specific neoantigens directly from human melanoma tissues. By constructing customized, patient-specific protein databases, they successfully mapped MS/MS spectra to somatic mutations. The study definitively proved that integrating mass spectrometry with genomic data significantly reduces the false-positive rates of in silico predictions, leading to the discovery of physically validated neoepitopes capable of eliciting strong, tumor-specific T cell responses.
Methods
This study highlights a rigorous proteogenomic pipeline to physically validate algorithmic predictions using actual patient tumor samples.
Key Technical Features:
- Sample types: Patient-derived native melanoma tissues and matched healthy cells.
- Custom Database Generation: WES and RNA-seq were utilized to translate somatic point mutations and indels into customized patient-specific FASTA databases.
- Immunopeptidomics: HLA class I complexes were purified via immunoaffinity, and peptides were eluted for deep analysis using Orbitrap mass spectrometers.
- Stringent Filtration: Candidates were filtered based on strict FDR control, transcriptomic expression support, and algorithmic binding predictions (NetMHCpan).
- Immunogenicity Tests: Validated neoantigens were co-cultured with patient-derived tumor-infiltrating lymphocytes (TILs) to confirm T cell reactivity.
To support research following these advanced proteogenomic principles, Creative Proteomics offers specialized neoantigen discovery and validation services featuring:
- Custom variant database construction from customer WES/RNA-seq data.
- High-resolution HLA immunopeptidomics profiling.
- Multi-layer evidence integration to filter false positives.
- Ranked candidate reporting for downstream functional assays and personalized vaccine design.
Results
Mass Spectrometry Validates True Presentation
- Out of thousands of computationally predicted somatic mutations, only a highly specific subset of mutated peptides was physically detected by mass spectrometry on the tumor cell surface.
Discovery of Highly Immunogenic Targets
- The proteogenomic approach successfully identified novel mutant neoepitopes (e.g., derived from NCAPG2 and SYTL4 genes) that were completely absent in matched normal tissues.
Functional T-Cell Activation Confirmed
- T-cell assays confirmed that the MS-validated neoepitopes successfully triggered robust interferon-gamma (IFN-γ) secretion from the patient's own CD8+ T cells, proving their viability as clinical immunotherapy targets.
Reference
- Bassani-Sternberg, M., Bräunlein, E., Klar, R., et al. "Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry." Nature Communications 7, 13404 (2016). DOI: 10.1038/ncomms13404
Frequently Asked Questions
What is the difference between neoantigen prediction and validation? +
Prediction relies on genomic sequencing (WES/RNA-seq) and computer algorithms to guess which mutated peptides might bind to an HLA molecule. Validation utilizes immunopeptidomics (mass spectrometry) to physically extract and measure the peptides from the tumor tissue, definitively proving that the predicted mutation is actively processed and presented on the cell surface.

Can you validate candidates using my existing WES and RNA-seq FastQ data? +
Yes. If your bioinformatics team has already generated sequencing data, we can securely ingest your FastQ, BAM, or VCF files. We will use your data to construct the customized variant search database, allowing us to focus our efforts entirely on the physical MS-assisted validation step, saving you time and sequencing costs.
How do you reduce the false discovery rate of computational algorithms? +
Algorithms often yield >90% false-positive rates due to biological processing bottlenecks. We reduce this by enforcing an intersection of evidence: candidates must have transcriptomic expression support (RNA-seq TPM thresholds) and strict physical detection via MS/MS. We employ customized decoy databases to maintain a <1% FDR during spectral matching.
Do you support both HLA Class I and Class II validation? +
Yes. Our immunopeptidomics pipelines utilize specific antibodies to independently enrich for both HLA Class I (CD8+ T cell targets) and HLA Class II (CD4+ T cell targets) complexes, allowing for comprehensive validation of the entire tumor ligandome.
What if I don't have matched normal tissue available? +
Matched normal tissue (or blood/PBMC) is highly recommended to accurately filter out patient-specific germline variations. If unavailable, we can perform tumor-only sequencing and filter against massive population databases (like dbSNP). However, this slightly increases the risk that a small fraction of validated targets may actually be private germline variants rather than true somatic neoantigens.
Can your pipeline identify fusions and cryptic non-canonical peptides? +
Absolutely. Because our customized databases integrate deep RNA-seq data, we can perform 6-frame translations to detect and physically validate neoantigens arising from gene fusions, retained introns, and transcriptionally active non-coding regions that standard WES algorithms overlook.
What happens after validation? Do you support downstream functional assays? +
Once the top targets are validated and ranked by our pipeline, we can synthesize the shortlisted peptides to support your internal functional workflows, or you can leverage our extended bioanalytical platforms (such as SPR or BLI) to further characterize TCR-peptide-MHC binding kinetics.