From Cryptic BGC to Structurally Validated Peptide: Closing the Discovery Gap
A single Streptomyces genome may contain 30–50 biosynthetic gene clusters (BGCs), yet the majority resist expression under standard laboratory conditions. When compounds are produced, the same known scaffolds — streptothricins, surfactins, actinomycins — dominate the LC-MS profile, masking low-abundance novel species. Traditional workflows rely on bioactivity-guided fractionation, which is labour-intensive and biased toward highly abundant or previously characterised metabolites. The consequence is a high rediscovery rate and a substantial investment of time and material on each candidate before structural information is available.
This service addresses the problem by integrating genome mining, high-resolution mass spectrometry, and automated dereplication into a single pipeline. Rather than proceeding blind, each project begins with a computational assessment of the strain's biosynthetic potential, allowing the analytical method to be tailored to the predicted peptide classes. This front-loaded strategy reduces the number of isolates requiring full characterisation and increases the probability that each candidate represents a genuinely new structure.
What the Service Covers: From Genome Scan to Peptide Structure
Six integrated modules span the complete discovery workflow, from initial genome annotation through to targeted quantification of validated candidates.
Peptide Classes and Biosynthetic Origins Accommodated by the Platform
The platform accommodates all major classes of microbially encoded peptides. Representative compound families and the analytical approach applied to each are listed below.
| Peptide Class | Representative Examples | Key Structural Features | MS Approach |
|---|---|---|---|
| Class I Lanthipeptides | Nisin, mersacidin | Lanthionine / methyllanthionine bridges; dehydrated residues (Dha, Dhb) | HRMS + HCD/ETD |
| Class II/III Lanthipeptides | Lacticin 3147, catenulipeptin | Multi-ring lanthionine topology; labionin bridges | HRMS + UVPD/SID |
| Thiopeptides | Thiostrepton, berninamycin | Thiazole/oxazole heterocycles; macrocyclic scaffold | HRMS + CID MS3 |
| Sactipeptides | Thurincin H, subtilosin A | Cysteine thioether crosslinks to α-carbons | HRMS + ETD |
| Bottromycins | Bottromycin A2 | Macrolactam; β-methylated amino acids | HRMS + HCD |
| Linear Non-Ribosomal Peptides (NRPs) | Gramicidin S, tyrocidine | Non-proteinogenic monomers; D-amino acids; halogenations | HRMS + data-dependent MS/MS |
| Cyclic Lipopeptides | Daptomycin, fengycin | Fatty acid tail; macrolactone ring | HRMS + RPLC gradient optimisation |
| Peptide-Polyketide Hybrids | Epothilone, bleomycin | Mixed NRP/PKS assembly; complex tailoring | HRMS + spectral network analysis |
| Fungal Cyclopeptides | Cyclosporin, beauvericins | N-methylated residues; macrocyclic; hydrophobic | HRMS + HILIC-MS |
| Diketopiperazines (DKPs) | Brevianamides, asperthecin | 2,5-diketopiperazine core; prenylated variants | HRMS + CID low-energy |
Analytical Technology Supporting Deep Structural Resolution
The platform combines three mass spectrometry systems and five fragmentation modes, each serving a distinct role in the discovery workflow. Genome-predicted structural complexity drives instrument and acquisition mode selection per project.
Technical Highlights
- Orbitrap Fusion Lumos — Sub-2 ppm Full-Scan HRMS
Parallel reaction monitoring (PRM) and data-dependent acquisition across a dynamic range exceeding 5 orders of magnitude; essential for confident sum formula assignment at <2 ppm deviation. - UVPD (Ultraviolet Photodissociation, 193 nm)
Preserves labile PTMs during fragmentation — critical for lanthipeptide ring topology determination, D-amino acid assignment, and N-methylation localisation not resolved by standard CID alone. - Ion Mobility–MS (TIMS-TOF Pro)
Separates isobaric peptide variants by collision cross-section (CCS), resolving co-eluting structural isomers and identifying macrocyclic scaffolds that overlap in retention time and nominal mass. - GNPS Molecular Networking
Spectral similarity clustering maps fragmentation relationships across the entire extract; isolated network nodes not clustering with any known compound family are flagged as priority de novo targets. - BiG-SCAPE / CORASON BGC Networking
Genome-level clustering of BGCs across a strain collection provides novelty-rank scores, directing MS acquisition toward producers with the most biosynthetically unique clusters. - Three-Tier Structural Confidence System
Results are reported as putative (exact mass + database negative), probable (mass + MS/MS ion series), or confirmed (mass + MS/MS + BGC precursor sequence match + PTM localisation).
 Orbitrap Fusion Lumos Tribrid
Orbitrap Fusion Lumos Tribrid
(Fig from Thermo Scientific)
 Agilent 6550 iFunnel Q-TOF
Agilent 6550 iFunnel Q-TOF
(Fig from Agilent Technologies)
 Bruker timsTOF Pro
Bruker timsTOF Pro
(Fig from Bruker)
Instrument Capability Overview
| Feature | Orbitrap Fusion Lumos | Agilent 6550 Q-TOF | Bruker timsTOF Pro |
|---|---|---|---|
| Mass Accuracy | <2 ppm RMS | <2 ppm (IRM-corrected) | <5 ppm |
| Fragmentation Modes | CID, HCD, ETD, EThcD, UVPD | CID (stepped collision energy) | CID (PASEF) |
| Ion Mobility | — | — | TIMS (CCS measurement) |
| Primary Application | De novo sequencing; PTM mapping; PRM quantification | Untargeted profiling; sum formula confirmation | Structural isomer resolution; high-throughput screening |
Why This Approach Reduces Rediscovery and Accelerates Structural Assignment
End-to-End Workflow: Six Stages from Genome Sequence to Characterised Candidate
Genome Submission & BGC Prioritisation
antiSMASH, PRISM, and BAGEL4 annotate all peptide-encoding BGCs; BiG-SCAPE clusters BGCs across the collection and ranks by novelty score.
Fermentation Condition Optimisation
OSMAC screening across 4–8 media conditions at two time points; epigenetic elicitors evaluated for cryptic BGC activation; SPE with mixed-mode cartridges for broad-polarity recovery.
UPLC-HRMS/MS Data Acquisition
Full-scan DDA on Orbitrap Fusion Lumos (m/z 150–2000, R = 120,000); top-20 precursor selection with HCD and parallel ETD/UVPD for BGC-matching candidates.
Dereplication & Novel Feature Identification
GNPS molecular networking clusters spectra by cosine similarity; isolated nodes and low-annotation clusters retained as novel candidates; cross-checked against DNP, AntiMarin, and in-house library.
De Novo Sequencing & Structural Assignment
MS3 and UVPD resolve lanthionine bridges, D-residue positions, and N-methylation; sequence validated against genome-predicted precursor; results reported in three confidence tiers.
Targeted Quantification & Reporting
MRM/PRM transitions developed and validated (linearity, LOQ, recovery); quantification applied across fermentation time points; final report includes annotated spectra, BGC-to-product tables, and raw data files.
1
Genome Submission & BGC Prioritisation
Client provides draft or complete genome sequence(s) or raw WGS reads. antiSMASH, PRISM, and BAGEL4 annotate all peptide-encoding BGCs with predicted precursor peptides, core motifs, and tailoring enzyme families. BiG-SCAPE clustering across the strain collection ranks BGCs by novelty score, directing resources toward producers with the most biosynthetically distinct clusters. Clients can provide FASTA genome files or raw Illumina FASTQ reads (≥5× coverage) for in-house assembly.
2
Fermentation Condition Optimisation
OSMAC strategy screens 4–8 media conditions in parallel (ISP2, R2A, YEME, and defined minimal media) at early and late stationary phases. Epigenetic elicitors (sodium butyrate, 5-azacytidine) are evaluated where cryptic BGC expression is predicted. SPE with mixed-mode cartridges (reversed-phase/ion-exchange) maximises recovery across the polarity range relevant to the predicted BGC compound class.
3
UPLC-HRMS/MS Data Acquisition
Crude extracts are analysed by UPLC (C18 reversed-phase, 1.7 µm, 2.1 × 100 mm column) coupled to Orbitrap Fusion Lumos in full-scan mode (m/z 150–2000, R = 120,000 at 200 m/z). Data-dependent acquisition selects the top 20 precursors per cycle for HCD fragmentation; candidate ions matching predicted BGC masses receive parallel ETD or UVPD fragmentation for PTM localisation. timsTOF TIMS data are acquired in parallel for projects requiring structural isomer resolution.
4
Dereplication & Novel Feature Identification
GNPS molecular networking clusters all MS/MS spectra by cosine similarity (threshold 0.65, minimum 6 shared peaks). Features clustering with known natural product families are flagged as known. Isolated network nodes and nodes in clusters with <3 known members are retained as novel candidates. Cross-referencing against DNP, AntiMarin, and the in-house spectral library provides a secondary dereplication layer, achieving combined known-compound flagging rates of >90%.
5
De Novo Sequencing & Structural Assignment
Priority candidates undergo multi-stage MS/MS (MS3) and, where needed, UVPD fragmentation to resolve lanthionine bridges, D-residue positions, and N-methylation patterns. The deduced sequence is validated against the genome-encoded precursor peptide. Molecular formula is confirmed by exact mass measurement (<2 ppm deviation) with isotope pattern analysis. Results are compiled with three confidence tiers: putative (mass + dereplication-negative), probable (mass + MS/MS ion series), or confirmed (mass + MS/MS + BGC precursor sequence match + PTM localisation).
6
Targeted Quantification & Reporting
For confirmed or probable candidates, MRM or PRM transitions are developed and validated (linearity, LOQ, inter-day precision, recovery). Quantification is applied across fermentation time points or fractionation stages to support titre optimisation. The final report includes raw data files (mzML, .raw), annotated spectra, candidate structure summaries, BGC-to-product correlation tables, quantification data tables, and recommended next steps for each validated candidate.
Sample Submission Guidelines
| Sample Type | Minimum Input | Recommended Format | Storage & Shipping |
|---|---|---|---|
| Fermentation broth (crude) | 50 mL per condition | Centrifuge-clarified supernatant; filter-sterilised (0.22 µm) | −80 °C; dry ice shipping |
| Freeze-dried extract | 5 mg per sample | Dissolved in DMSO or MeOH at ≤10 mg/mL | −20 °C; ambient shipping acceptable for ≤72 h |
| Bacterial cell paste (whole-cell extract) | 200 mg wet weight | Flash-frozen pellet; do not add lysis buffer | −80 °C; dry ice |
| Fungal mycelial mat | 500 mg wet weight | Flash-frozen; excess agar removed | −80 °C; dry ice |
| Purified fraction (semi-pure) | 0.5 mg lyophilised | Reconstitute in 50% MeOH/H2O; avoid TFA >0.1% | −20 °C; ambient acceptable |
| Genomic DNA / WGS reads (genome mining only) | ≥100 ng DNA or FASTQ file | FASTQ (paired-end, ≥5× genome coverage); high-MW gDNA preferred | Ambient (DNA); secure upload (FASTQ) |
Notes: Samples containing >5% DMSO may suppress ionisation; dilute prior to submission. Avoid sodium azide or detergents (SDS, Triton) in buffers. A minimum of 3 biological replicates per condition is recommended for statistically valid differential analysis. Contact our project team for custom guidelines for anaerobic microbial cultures.
Representative Results from Microbial Peptide Discovery Projects
GNPS Dereplication Network — Novel Nodes from a Streptomyces sp. Extract
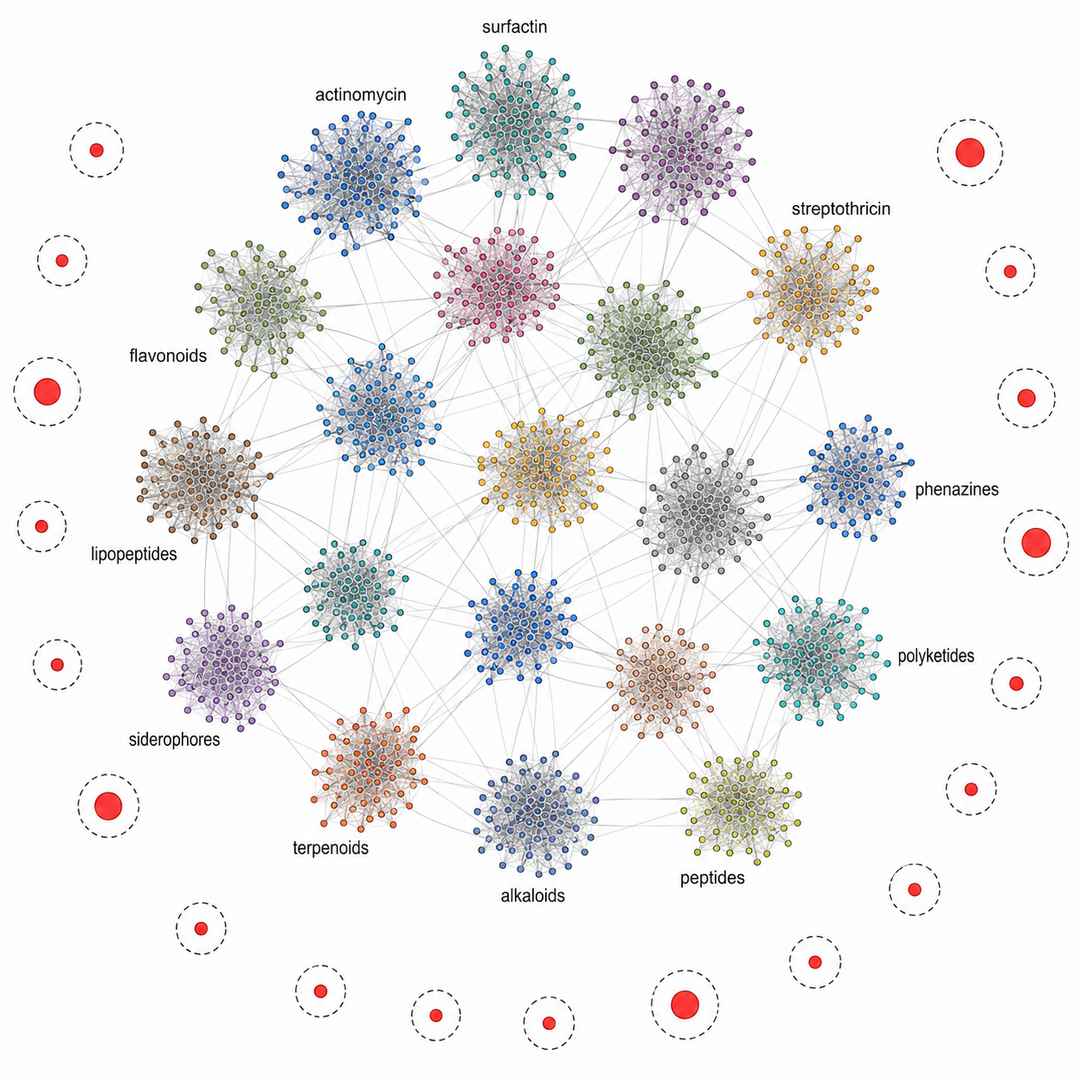
GNPS molecular network from a Streptomyces sp. ISP-5 extract screened across 10 media conditions. Known compound families form 18 annotated clusters; 14 isolated nodes match antiSMASH-predicted Class II lanthipeptide BGC masses and return no database hits, flagging them as priority de novo candidates.
UVPD Fragmentation — Lanthionine Bridge Topology in a 3.4 kDa RiPP Candidate
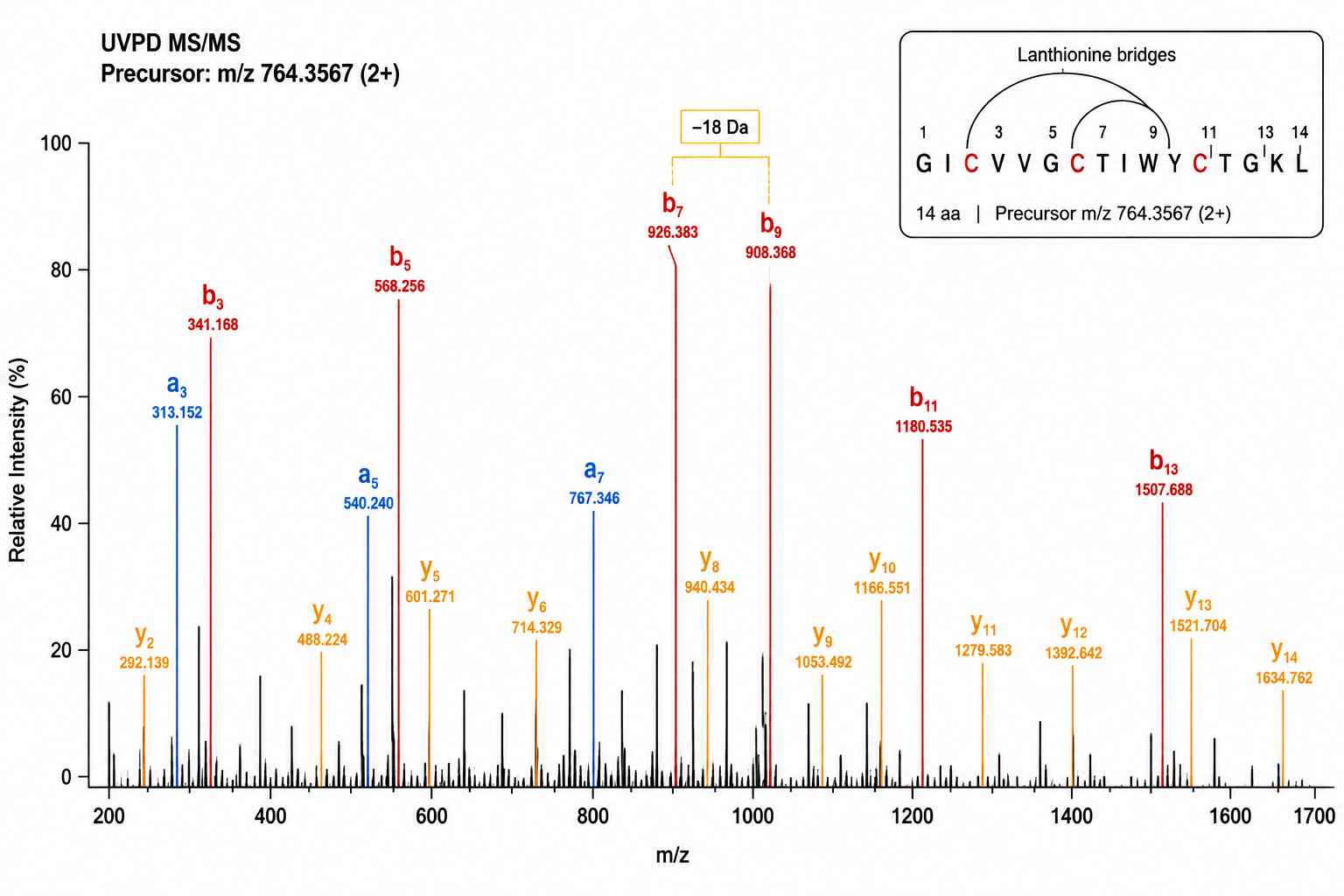
UVPD spectrum of the [M+4H]4+ precursor at m/z 851.42 provides 94% sequence coverage. A −18 Da gap at positions 7–9 confirms dehydration at Ser7; a crosslink-induced fragment localises the first lanthionine bridge between Cys3 and Cys9, consistent with the genome-predicted core peptide.
MRM Fermentation Time-Course — Cyclic Lipopeptide Titre in Bacillus subtilis
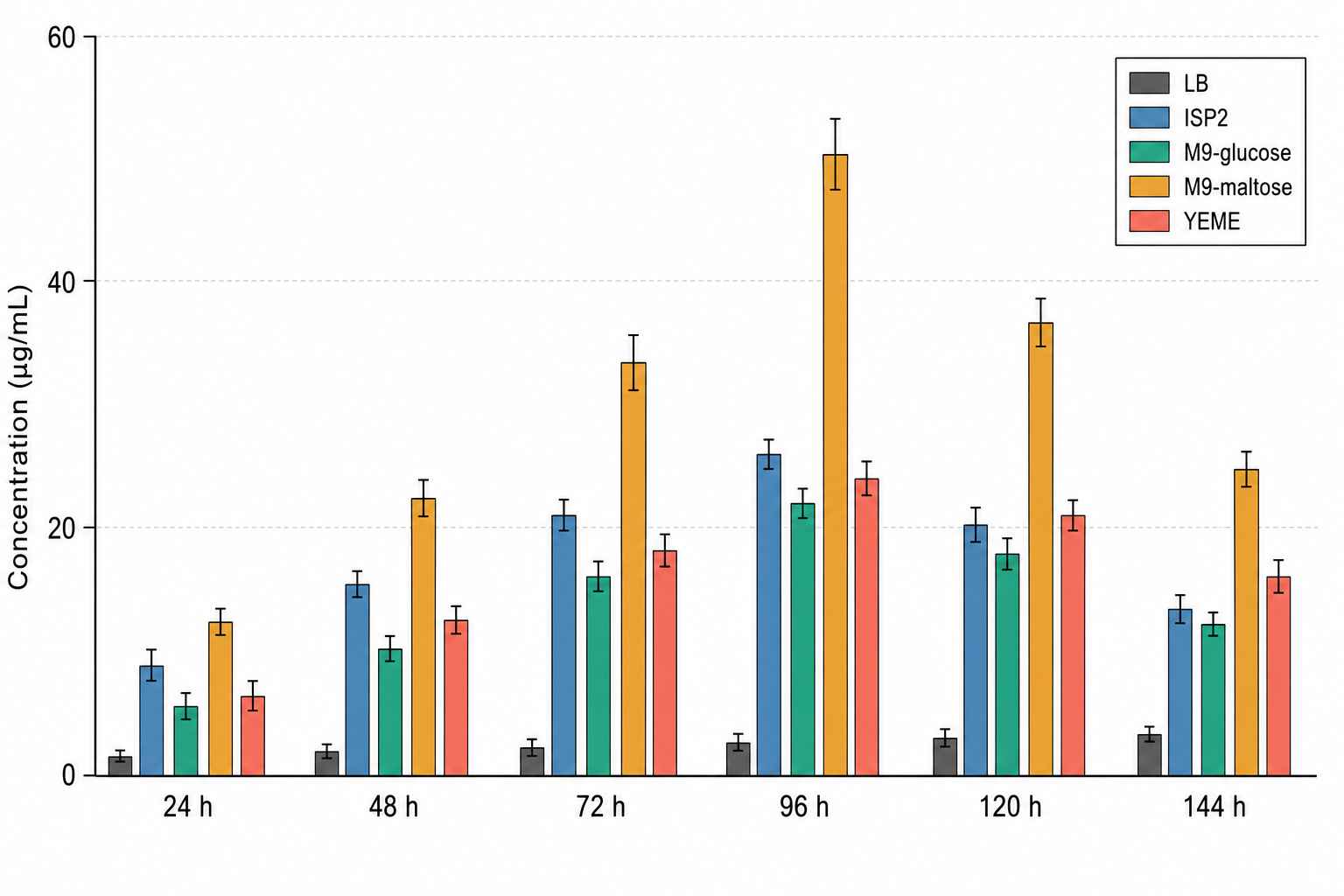
MRM quantification of a cyclic lipopeptide across a 144 h fermentation in Bacillus subtilis ATCC 6633. Peak titre reached 48.7 ± 3.2 µg/mL at 96 h in M9-maltose medium, versus ≤3 µg/mL in LB despite higher biomass — directly informing scale-up condition selection.
BGC-to-Product Correlation — 24-Strain Marine Actinomycete Screen
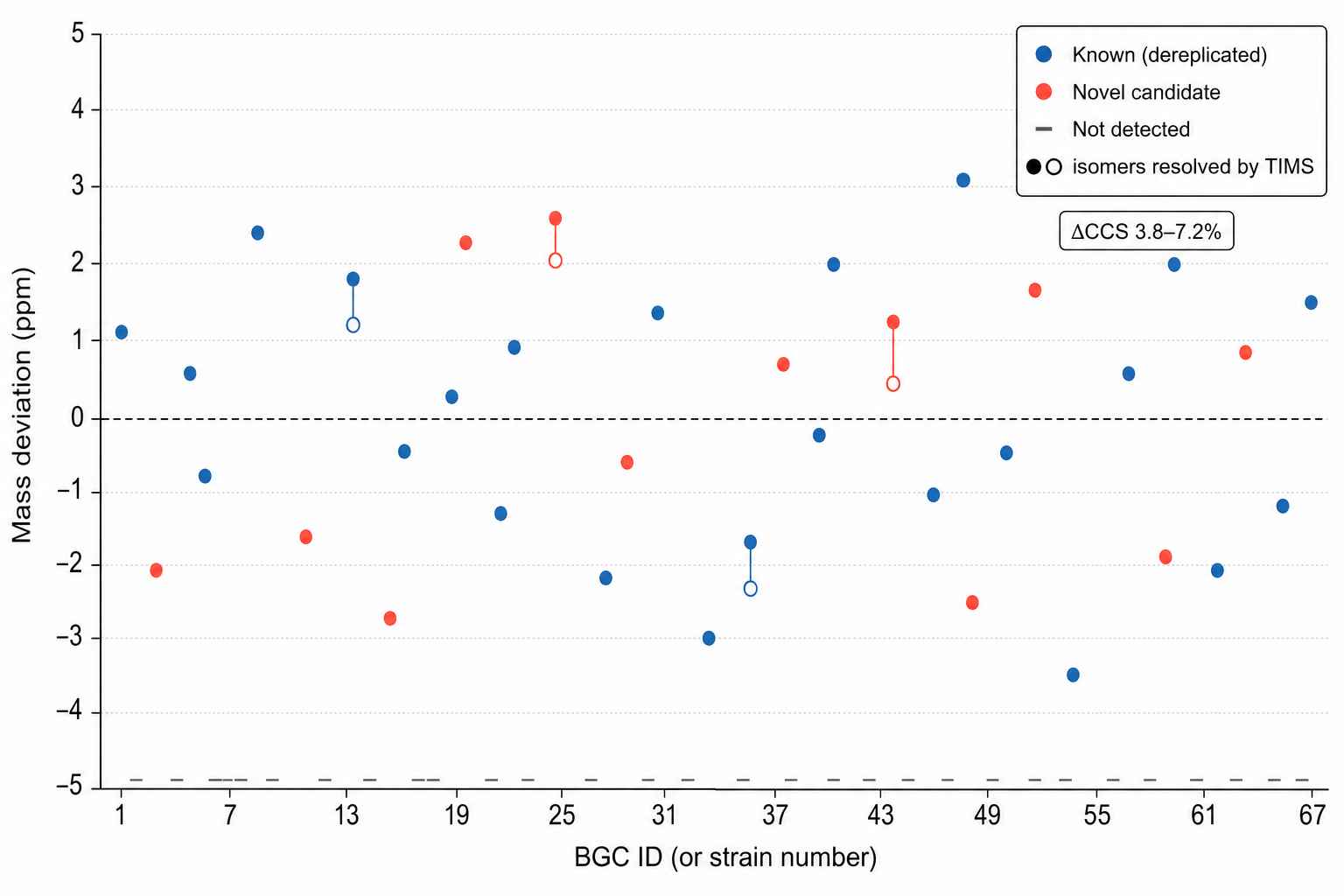
Across 67 antiSMASH-predicted peptide BGCs, genome-guided MS detected 41 matching precursor ions (61% detection rate); 13 were flagged as novel after dereplication. TIMS ion mobility resolved 4 isobaric pairs by CCS, identifying structural isomers indistinguishable by retention time or nominal mass.
Research Applications
- Antibiotic lead discovery: Identify novel peptide scaffolds from under-explored actinomycetes, marine bacteria, and fungal sources with structural features distinct from existing antibiotic classes.
- BGC activation studies: Monitor the metabolic response to epigenetic elicitors, co-cultivation conditions, or regulatory gene overexpression to determine which cryptic BGCs are activated.
- Heterologous expression validation: Confirm production and structural integrity of heterologously expressed peptides in chassis organisms (e.g. Streptomyces coelicolor M1152, Bacillus subtilis 168).
- Strain collection dereplication: Screen large microbial libraries to remove producers of known compounds before investing in bioassay-based prioritisation.
- Structure-activity relationship (SAR) support: Quantify the impact of biosynthetic enzyme mutants or feeding experiments on the ratio of modified to unmodified peptide species.
- Regulatory metabolomics: Characterise peptide secondary metabolite profiles as a readout of regulatory circuit activity (e.g. quorum sensing, σ-factor mutants).
- Environmental metagenomics: Link metagenomic BGC inventories to detected metabolites in environmental extracts (soil, marine sediment, gut microbiome fractions).
- Fermentation process development: Provide quantitative titre data across process variables (pH, temperature, dissolved oxygen, carbon source) to guide early-stage bioprocess optimisation.
Deliverables
- Genome mining report: annotated BGC list with predicted compound class, precursor peptide sequence, tailoring enzymes, and novelty rank score
- Fermentation condition screen summary: extract yield, UV absorbance profiles, and preliminary TIC comparison across all tested conditions
- Full-scan HRMS datasets in open mzML format (compatible with MZmine, XCMS, Progenesis)
- GNPS molecular network files (.graphml) with annotation layers for known compound families and novel candidate nodes
- Dereplication report: compound match list with database source, spectral similarity score, and known/novel classification
- De novo sequencing reports for each priority candidate: annotated MS/MS spectra, proposed sequence, PTM assignments, and confidence tier (putative / probable / confirmed)
- BGC-to-product correlation table with precursor mass accuracy, ion series coverage, and genetic evidence summary
- MRM/PRM quantification data: calibration curves, LOD/LOQ, inter-day precision, and sample concentration tables (where quantification is in scope)
- Ion mobility CCS values for all structural candidates (timsTOF projects only)
- Final integrated report (PDF) with executive summary, methods, results, and recommended next steps for each validated candidate
Dedicated Microbial Peptide Discovery vs. General Metabolomics: Key Differences
| Dimension | General Untargeted Metabolomics | Dedicated Microbial NP Peptide Discovery (This Service) |
|---|---|---|
| Database Coverage for Dereplication | HMDB, METLIN, MassBank — optimised for primary metabolites and mammalian lipids; limited NP peptide entries | GNPS NP spectral library, AntiMarin, DNP, in-house RiPP/NRP library (>8,000 entries); purpose-built for secondary metabolite dereplication |
| Fragmentation Strategy | CID/HCD only; b/y-ion series; optimised for tryptic peptides with standard amino acids | CID + HCD + ETD + UVPD (193 nm); resolves lanthionine bridges, D-amino acids, NRP monomers, and N-methylation not covered by standard CID alone |
| Genome Integration | No genomic input; metabolites annotated post-hoc against chemical databases only | BGC predictions guide MS acquisition targets; each MS feature cross-referenced against predicted precursor masses from antiSMASH/BAGEL annotation |
| Rediscovery Risk Management | High; known scaffolds and novel compounds receive equal analytical priority; no active dereplication before data interpretation | Multi-layer dereplication (>90% known-compound flagging) before de novo interpretation; resources concentrated on genuinely unannotated features |
| Structural Confidence for Novel Hits | Putative annotation based on database match; no sequence-level evidence; no PTM localisation | Three-tier confidence system (putative / probable / confirmed): BGC sequence match + exact mass + MS/MS ion series coverage + PTM localisation |
| Quantification Capability | Relative quantification only; no compound-specific MRM/PRM method development | Absolute quantification via MRM/PRM with method validation (linearity, LOQ, recovery) for confirmed candidates |
What genome quality is required for the genome mining step? +
Draft assemblies with N50 >50 kb are sufficient for antiSMASH and BAGEL4 annotation. Complete circular genomes provide the best BGC boundary resolution, but the analytical phase can begin with high-quality draft assemblies. We accept raw Illumina short-read FASTQ files (≥5× coverage) for in-house de novo assembly, or completed assemblies in FASTA format.
Can the service process metagenomic samples rather than pure cultures? +
Yes. For environmental samples (soil, marine sediment, gut microbiome), GNPS-based spectral networking can be applied without a matched genome, or assembled metagenome BGC inventories can be integrated with metabolite detection. The genome-to-product correlation confidence is lower in metagenomic contexts, but de novo sequencing of detected peptides remains fully applicable.
How is "novel" defined in the dereplication output? +
A feature is classified as novel if it returns no spectral match above cosine similarity 0.75 in GNPS, DNP, AntiMarin, and the in-house library, AND its exact mass does not correspond to any known compound entry within 5 ppm. Compounds that match known families at the network level but differ by one or more modification sites are reported as analogues rather than novel entities, with the modification site annotated.
What is the detection limit for low-abundance peptides in fermentation extracts? +
On the Orbitrap Fusion Lumos in full-scan mode, the practical detection limit for peptides of 1–5 kDa is approximately 1–10 ng/mL in clarified fermentation broth, depending on ionisation efficiency and matrix complexity. For targeted detection of a predicted BGC product, PRM acquisition can lower this to <0.5 ng/mL. SPE concentration (typically 10–50× enrichment) is applied to all crude broth submissions before analysis.
Are D-amino acids and N-methylated residues detectable by this platform? +
Yes. D-amino acid configuration is inferred from UVPD fragmentation patterns (a/x ion series) and confirmed where necessary using Marfey's reagent derivatisation followed by LC-MS analysis. N-methylation is identified by the +14.016 Da mass shift per N-methyl group and confirmed by UVPD a-type ions. Both features are routinely encountered in NRP substrates such as cyclosporins, gramicidins, and lipopeptides.
Genome-Guided Discovery of Novel Non-Ribosomal Peptides via Integrated Genomics and LC-MS/MS
Journal: Nature Communications
Published: 2021
Behsaz B, et al. Integrating genomics and metabolomics for scalable non-ribosomal peptide discovery. Nat Commun. 2021;12:3225. DOI: 10.1038/s41467-021-23502-4
Background
Non-ribosomal peptides (NRPs) are among the most structurally diverse class of microbial natural products, encompassing antibiotics, antifungals, immunosuppressants, and cytotoxic agents. They are assembled by large modular enzyme complexes (NRPSs) and frequently incorporate D-amino acids, N-methylated residues, β-hydroxy acids, and macrolactone or macrolactam ring systems — features that make them resistant to standard proteomics-based detection. Genome sequencing has revealed that most microbial producers carry far more NRP biosynthetic gene clusters (BGCs) than metabolites ever detected in the laboratory, a gap attributed to cryptic expression and the limitations of untargeted metabolomics workflows not anchored to the genetic context.
Research Challenge
The central problem the study addressed is directly relevant to clients exploring novel NRP space: when an extract is analysed by LC-MS/MS without genomic guidance, the majority of MS/MS spectra remain unannotated, and genuinely new NRP scaffolds are indistinguishable from the noise of the uncharacterised metabolome. Standard GNPS networking clusters spectra by similarity but does not link them to BGC predictions, leaving the question of biosynthetic origin unresolved. Conversely, antiSMASH BGC annotation provides rich genetic information but no direct mass spectral validation. The absence of a scalable method to bridge these two data streams meant that, for most projects, genuine novel NRPs could only be found by labour-intensive iterative fractionation and NMR — a throughput bottleneck incompatible with strain collection screening.
Analytical Approach
The NRPminer tool was applied to three microbial datasets spanning 155 Xenorhabdus/Photorhabdus genomes, a Bacillus collection, and a published Streptomyces set. The workflow closely mirrors the service pipeline offered here:
- BGC annotation: antiSMASH was used to annotate all NRP-encoding BGCs in each genome, generating predicted precursor amino acid sequences and tailoring modification profiles for each cluster.
- Theoretical spectral library construction: Predicted NRP sequences were converted to theoretical MS/MS fragment libraries, accounting for D-amino acid configurations, N-methylation, epimerisation, β-methylation, and macrocyclisation — a modification-tolerant framework rather than a fixed database lookup.
- LC-MS/MS acquisition and GNPS networking: Extract spectra were clustered by cosine similarity in GNPS; isolated nodes and low-annotation clusters were cross-referenced against the theoretical BGC-derived spectral library rather than only against known compound databases.
- BGC-to-product matching and scoring: NRPminer matched experimental MS/MS spectra to genome-predicted NRP fragments using a probabilistic scoring model, explicitly tolerating variable modifications. Candidate matches were ranked by spectral coverage and biosynthetic plausibility.
- NMR confirmation of priority candidates: The top novel candidates (protegomycin, xenoamicin-like family, aminformatide) were isolated and validated by NMR to confirm the spectral assignments.
Key Findings
In the XPF dataset alone, NRPminer identified 9 known NRP families and uncovered 3 previously uncharacterised NRP families. Protegomycin, a novel lipopeptide from Photorhabdus, was fully characterised by NMR and subsequently confirmed as structurally distinct from all existing NRP classes. Across all datasets, 180 NRPs were identified — the majority lacking prior database entries — demonstrating that genomic integration substantially expands what LC-MS/MS can detect compared to untargeted metabolomics alone. Critically, the approach was scalable: the entire computational workflow ran on standard hardware in hours, making it applicable to multi-strain screening projects rather than individual isolates.
What This Means for Your Project
This study illustrates the core principle behind the service: anchoring MS/MS acquisition and interpretation in BGC context transforms what is achievable in a single project cycle. Several specific outcomes are directly transferable to client projects:
- Rediscovery risk is quantifiable upfront. Before any LC-MS run, BGC novelty ranking identifies which strains carry clusters with no close relatives in public databases — concentrating analytical effort on the most productive starting material.
- Highly modified NRPs are no longer invisible. The modification-tolerant matching framework used here, and implemented in our service pipeline, recovers D-amino acid-containing, N-methylated, and macrocyclic NRPs that would score zero against standard proteomics databases.
- Novel compounds can be structurally assigned at the MS/MS level. The BGC-to-product correlation provides a structural hypothesis — sequence, modification sites, cyclisation pattern — before any NMR is performed, sharply reducing the fraction of the isolation effort that yields no structural information.
- Strain collection screening is tractable. The same computational pipeline scales from a single strain to hundreds, making it feasible to rank an entire collection by chemical novelty potential within one project.
Reference
- Behsaz B, Bode E, Gurevich A, et al. Integrating genomics and metabolomics for scalable non-ribosomal peptide discovery. Nat Commun. 2021;12:3225. https://doi.org/10.1038/s41467-021-23502-4