Why Disulfide Connectivity Defines DRP Bioactivity
Disulfide-rich peptides (DRPs) are a structurally distinct class of bioactive molecules constrained by two or more disulfide bonds, forming rigid, folded scaffolds that confer exceptional proteolytic resistance, target affinity, and serum stability. Examples span ω-conotoxins (voltage-gated calcium channel blockers), cyclotides (macrocyclic knottins such as kalata B1), knottins (cystine-knot miniproteins), spider toxins (vanillotoxins, hanatoxin), and plant defensins. The defining feature of DRPs — their disulfide connectivity — directly determines the three-dimensional geometry of the bioactive surface, and therefore receptor binding affinity and selectivity.
A critical challenge distinguishes DRP characterisation from standard peptide analysis: mass alone cannot confirm bioactivity. A conotoxin with three disulfide bonds has 15 possible connectivity isomers (Cys pairing arrangements), each with an identical monoisotopic mass. Standard reversed-phase HPLC separates by hydrophobicity, not disulfide topology — correctly folded and misfolded isoforms frequently co-elute. Reduced LC-MS, which breaks all disulfide bonds, reports only the total cysteine count and provides no connectivity information whatsoever.
The structural biology literature makes this distinction stark. A 2020 study of a novel T1-conotoxin isolated from Conus bandanus required a combination of MALDI-TOF mass profiling and ETD/CID tandem MS to resolve the connectivity of four cysteine residues — revealing a non-standard pairing that was essential to the peptide's pharmacological activity (Bao et al., 2020). Without ETD-based fragmentation under non-reducing conditions, this connectivity would have been misassigned.
The non-reducing LC-MS + ETD/ECD workflow exists precisely because standard analytical approaches cannot make this distinction. Native disulfide bonds are preserved during ionisation; electron-transfer dissociation then cleaves the peptide backbone while leaving disulfide bonds intact, generating characteristic fragment ion pairs that directly reveal which cysteine residues are linked.
What We Offer: Disulfide Mapping, Oxidative Folding & Engineering Support
A comprehensive, end-to-end service for disulfide-rich peptide characterisation — from confirming oxidative folding state and assigning disulfide connectivity, to providing preliminary engineering consultation for stability and developability optimisation.
Detectable Disulfide-Rich Peptide Types
| Peptide Class | Typical Disulfide Topology | Common Source | Primary Analytical Challenge |
|---|---|---|---|
| α-Conotoxin | 2 disulfide bonds (CysI–CysIII, CysII–CysIV) | Conus snail venom | Discriminating native vs ribbon vs bead isomers; low pmol quantities |
| ω-Conotoxin | 3 disulfide bonds (6 Cys, 3 pairings) | Conus snail venom | 15 possible connectivity isomers; ETD required for unambiguous assignment |
| Cyclotide (knottin) | 3 disulfide bonds in cyclic cystine knot (CCK) | Plants (Viola, Rubiaceae) | Head-to-tail macrocycle + disulfide knot; non-reducing conditions essential |
| Knottin miniprotein | 3 disulfide bonds, cystine knot scaffold | Engineered / recombinant | Connectivity in engineered variants may deviate from natural fold |
| Spider toxin (vanillotoxin) | 2–4 disulfide bonds | Spider venom glands | Heterogeneous venom matrix; peptide enrichment before MS required |
| Plant defensin | 4 disulfide bonds (2 pairs) | Seeds, leaves | Cys-stabilised α-helix motif; multiple folding pathways |
| μ-Conotoxin | 3 disulfide bonds | Conus snail venom | Longer sequences (16–22 AA); disulfide vs. backbone heterogeneity |
| Bacteriocin (lantipeptide) | Thioether rings (lanthionine) | Bacterial culture | Lanthionine vs. conventional disulfide; requires specific fragmentation approach |
| Disulfide-rich cyclised peptide | 1–4 disulfide bonds + head-to-tail cyclisation | Engineered scaffolds | Cyclisation confirmation alongside connectivity; MSⁿ required |
| Miscellaneous knotted peptides | Variable (2–5 disulfide bonds) | Natural product libraries | Connectivity unknown a priori; iterative ETD + partial reduction workflow |
Non-Reducing vs. Reduced LC-MS: Why the Distinction Matters
The difference between reduced and non-reducing LC-MS is not merely technical — it determines whether disulfide connectivity information is preserved or permanently lost.
In reduced LC-MS, disulfide bonds are chemically broken (typically with dithiothreitol or TCEP) before analysis. The instrument detects individual cysteine-containing peptide fragments with a mass shift corresponding to a reduced sulfhydryl group. While this confirms the number and positions of cysteine residues, it provides zero information about which cysteines were originally paired. A conotoxin with 4 cysteines will show identical reduced-MS data whether it adopts the native fold or a completely scrambled isomer.
Non-reducing LC-MS maintains intact disulfide bonds throughout the analytical process. Under native electrospray ionisation conditions, peptide mass reflects the oxidised state. Fragmentation by ETD or ECD selectively cleaves the peptide backbone without reducing the disulfide bond, generating complementary b- and y-ions linked by the disulfide bridge. The mass difference between two disulfide-linked fragment ions directly reveals the cysteine pairing.
For peptides with multiple disulfide bonds, the non-reducing approach is the only way to determine connectivity. The partial reduction strategy — in which disulfide bonds are selectively reduced one pair at a time using controlled TCEP exposure — creates overlapping connectivity subsets that collectively resolve the entire disulfide network, even in highly constrained DRPs.
Platform Technology & Analytical Depth
The analytical platform integrates high-resolution Orbitrap mass spectrometry with non-reducing nanoLC separation and ETD/ECD fragmentation, optimised for disulfide-rich peptide characterisation across all scaffold types.
- Orbitrap-based HRMS with sub-ppm mass accuracy for monoisotopic mass confirmation
- ETD and ECD fragmentation modes — disulfide bonds remain intact during backbone cleavage, enabling direct connectivity assignment
- Non-reducing LC-MS workflow under native or mildly denaturing conditions; no DTT/TCEP pre-treatment that would destroy connectivity information
- Stepwise partial reduction with controlled TCEP exposure for complex multi-disulfide DRPs (≥3 disulfide bonds)
- PTM-aware fragmentation analysis: C-terminal amidation, pyroglutamylation, hydroxylation, and O-/N-linked glycosylation identified alongside disulfide mapping in a single run
- Capillary and nanoLC coupling for low-input peptide samples (≥50 µg minimum; dedicated μg-scale protocols available)
- Raw data archived and available on request; structured reports formatted for journal supplementary material and patent dossiers
| Analytical Capability | Creative Proteomics (DRP Platform) | Commercial Peptide Synthesis | General Analytical Facility |
|---|---|---|---|
| Disulfide Connectivity Assignment | ETD/ECD + partial reduction workflow | None (reduced MS only) | Limited (standard protease digestion) |
| Oxidative Folding State Confirmation | Non-reducing RP-HPLC + HRMS | None (HPLC purity only) | Rarely offered |
| Non-Reducing LC-MS Compatible | Yes — dedicated native workflow | No | Occasional; not specialised |
| ETD/ECD Fragmentation Available | Yes — Orbitrap platform | No | Variable |
| PTM + Disulfide Mapping (single run) | Yes | No | No |
| Low-Input DRP Characterisation (≤100 µg) | Yes — nanoLC-HRMS optimised | No | Sometimes |
Disulfide Mapping Workflow: From Non-Reducing MS to Connectivity Assignment
A five-step workflow integrating non-reducing LC-MS with ETD/ECD fragmentation to deliver connectivity maps and folding assessments for any disulfide-rich peptide scaffold.
Sample Receipt & Sequence Review
Peptide sequence, number of Cys residues, and available quantity confirmed; non-reducing sample preparation protocol selected
Non-Reducing LC-MS Characterisation
Intact mass confirmed under native conditions; oxidative folding heterogeneity assessed by RP-HPLC co-elution; MS1 accuracy verified before ETD
ETD/ECD Fragmentation & Connectivity Mapping
Disulfide-linked b- and y-ion pairs generated; cysteine pairings assigned; partial reduction applied for multi-disulfide DRPs
PTM Characterisation & Folding Report
Amidation, pyroglutamylation, and other PTMs documented alongside connectivity; folding homogeneity quantified
Engineering Consultation (Optional)
Preliminary assessment of oxidative stability, refolding recommendations, and cyclisation strategy for characterised hits
1
Sample Receipt & Sequence Review
Peptide sequence submitted by the client is reviewed for cysteine count, predicted disulfide framework, and net charge. Sample vial integrity and storage condition are inspected upon receipt. For crude or semi-purified material, an initial RP-HPLC purity check is performed before committing to the non-reducing workflow.
2
Non-Reducing LC-MS Characterisation
Intact peptide is analysed by nanoLC-HRMS under native, non-reducing conditions. Oxidative folding state is assessed by comparing the elution profile of the intact mass against a reduced control (run in parallel on a separate aliquot). MS1 mass accuracy is verified to be sub-ppm before proceeding to fragmentation.
3
ETD/ECD Fragmentation & Connectivity Mapping
The native (non-reduced) peptide is subjected to ETD or ECD fragmentation on an Orbitrap Fusion or timsTOF Pro. Backbone cleavage occurs while disulfide bonds remain intact, generating complementary b- and y-ion pairs linked by the disulfide bridge. The mass difference between linked ions directly reveals the cysteine pairing. For peptides with ≥3 disulfide bonds, a stepwise partial reduction protocol is applied to generate overlapping connectivity subsets.
4
PTM Characterisation & Folding Report
C-terminal amidation, N-terminal pyroglutamylation, hydroxylation, and other PTMs are identified alongside disulfide connectivity in the same analytical run. Folding homogeneity is quantified from the non-reducing RP-HPLC + HRMS overlay — the area ratio of correctly folded vs misfolded isomers is reported as the oxidative folding purity.
5
Engineering Consultation (Optional)
For characterised hits, a preliminary scientific consultation is available to discuss refolding strategies, cyclisation approaches, and proteolytic resistance improvements. The goal is to translate structural connectivity data into actionable developability guidance, accelerating the transition from a folded DRP hit to a structurally optimised lead candidate.
Sample Requirements
| Peptide Type | Minimum Amount | Preferred Format | Storage / Shipping | Special Notes |
|---|---|---|---|---|
| α-Conotoxin (synthetic, ≥2 Cys) | ≥ 50 µg | Lyophilized powder | −20 °C, dry ice | Include expected connectivity if known |
| ω-Conotoxin / μ-Conotoxin (≥3 Cys) | ≥ 100 µg | Lyophilized powder or 0.1% FA solution | −20 °C, dry ice | Partial reduction step requires adequate sample |
| Cyclotide (natural extract) | ≥ 100 µg | Solution in 0.1% FA or ACN/H₂O | 4 °C (short term); −20 °C (long term) | Confirm head-to-tail cyclisation by MS1 mass |
| Knottin miniprotein (recombinant) | ≥ 200 µg | Lyophilized or in solution | −80 °C | Buffer composition may affect oxidative folding protocol |
| Spider toxin (venom gland extract) | ≥ 200 µg (total peptide) | Crude venom or semi-purified fraction | −80 °C, dry ice | Enrichment step may be required; contact us first |
| Plant defensin | ≥ 100 µg | Lyophilized powder | −20 °C, dry ice | 4-disulfide topology; ETD optimised for larger peptides |
Representative Results
Non-Reducing LC-MS Profile of a 3-Disulfide Conotoxin
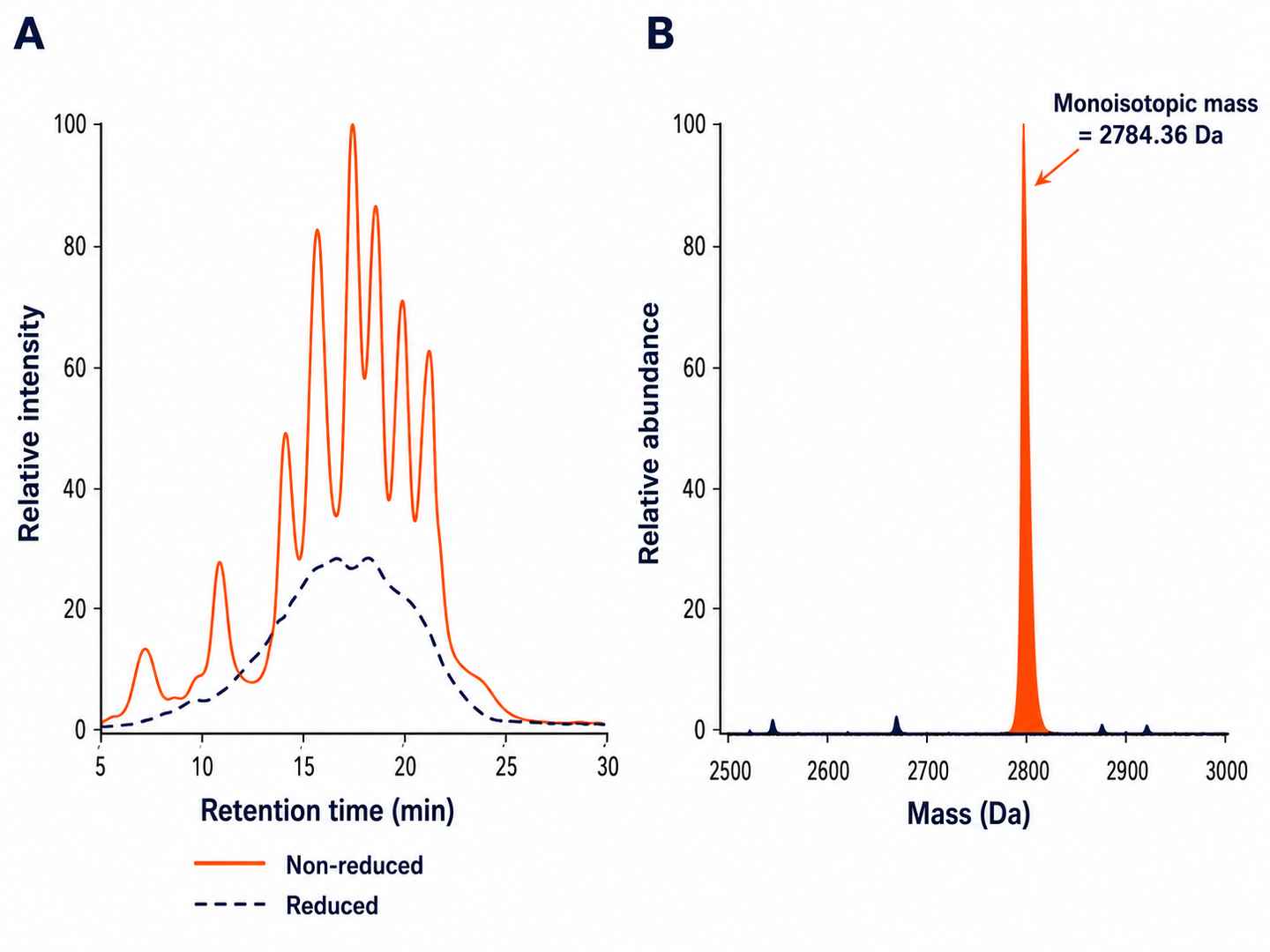
Overlaid total ion chromatogram (TIC) of reduced vs non-reduced conotoxin, with deconvoluted intact mass and isomer peak separation by nanoLC-HRMS. Folding heterogeneity is quantified from peak area integration.
ETD Fragment Ion Map for Disulfide Connectivity Assignment
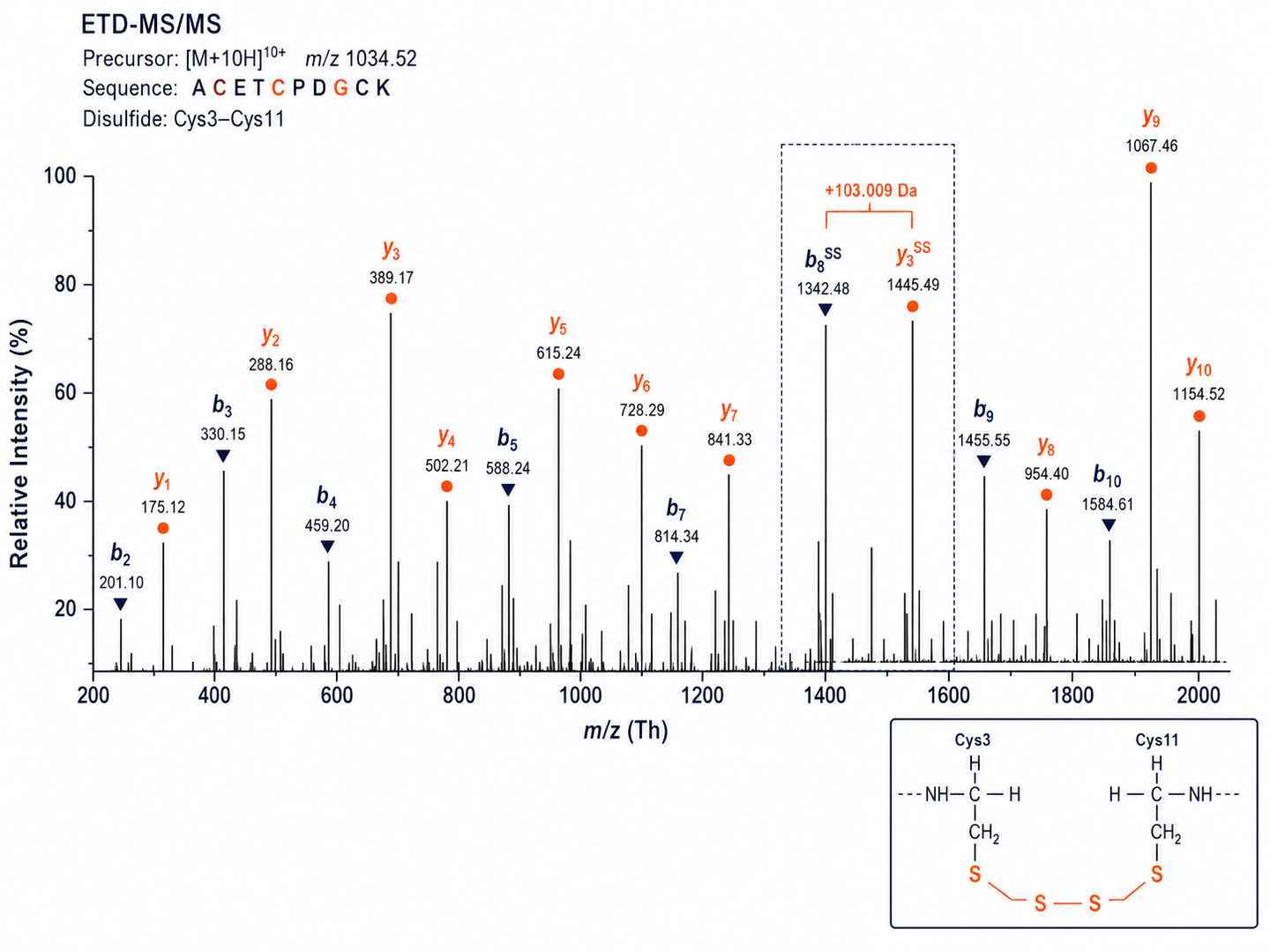
ETD-MS/MS spectrum with annotated b- and y-ions confirming specific Cys–Cys pairing. The mass difference between disulfide-linked fragment ion pairs directly reveals the cysteine connectivity for each pairing.
Oxidative Folding Purity Assessment by RP-HPLC + HRMS
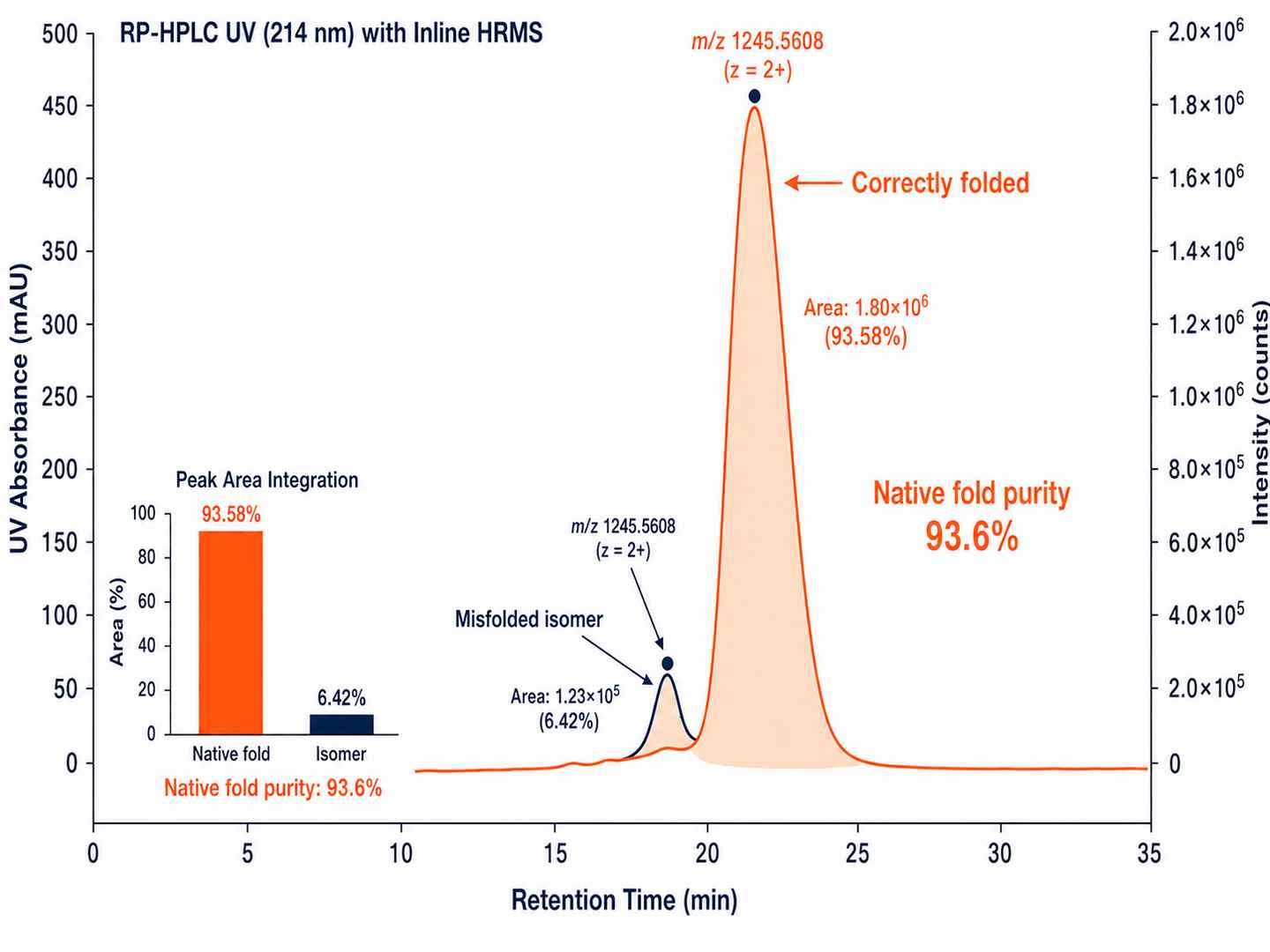
Co-elution of correctly folded vs misfolded isoforms monitored by RP-HPLC with inline HRMS confirmation. Oxidative folding purity is quantified and reported alongside the connectivity map.
Disulfide Mapping Heatmap Across Peptide Library
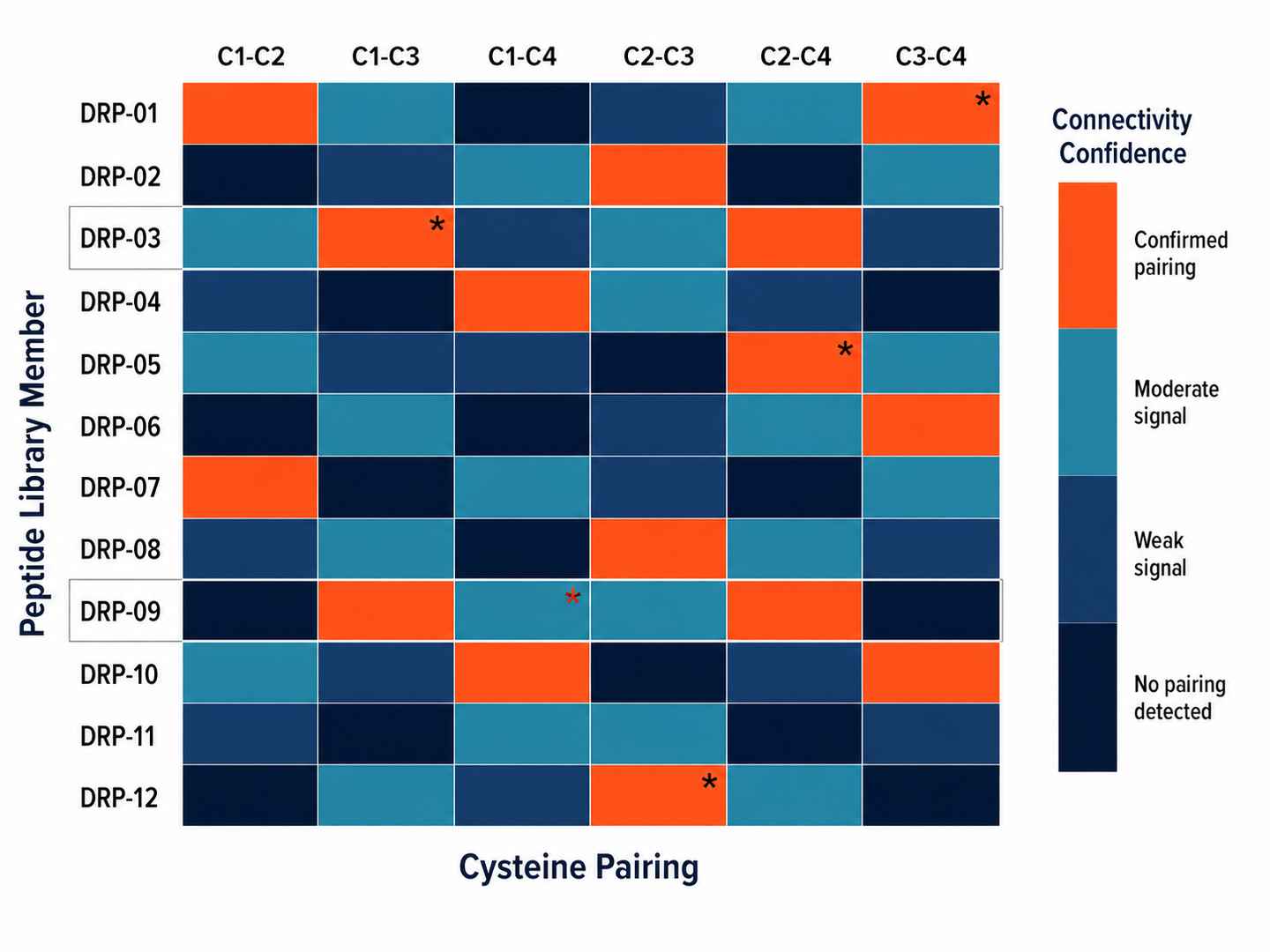
Heatmap showing predicted vs observed disulfide connectivity for 12 DRP library members. Mismatches are flagged with orange indicators, enabling rapid identification of misfolded or incorrectly paired library hits.
Engineering & Developability: From Structural Data to Better Peptides
- Serum stability and proteolytic resistance evaluation informed by disulfide scaffold rigidity
- Cyclisation strategy assessment: head-to-tail vs. side-chain-to-tail vs. stapled approaches
- Conformational constraint optimisation: which disulfide pairings to retain or modify for target selectivity
- Refolding protocol recommendations for misfolded DRP isomers identified during characterisation
- Lead peptide prioritisation based on oxidative folding homogeneity and structural integrity
- Crosslinking and immobilisation strategies for DRP conjugates used in therapeutic or diagnostic formats
- Comparative disulfide analysis across analogue series for structure-activity relationship (SAR) support
What's Included in the Deliverables
- Non-reducing intact mass spectrum with deconvoluted mass (monoisotopic and average)
- ETD/ECD fragmentation data with annotated connectivity map (b- and y-ion assignments)
- Partial reduction data for peptides with ≥3 disulfide bonds
- Oxidative folding homogeneity report (RP-HPLC peak areas + HRMS confirmation)
- PTM inventory with site-specific assignments where fragmentation permits
- Structured summary report in Word/PDF format, suitable for journal supplementary material and patent dossiers
- Raw instrument files (raw, mzML) archived and available on request
- Preliminary scientific consultation for characterised hits (optional add-on)
Why Choose Our Disulfide-Rich Peptide Characterisation Platform
Why can't standard HPLC or reduced LC-MS confirm disulfide connectivity? +
HPLC separates peptides by hydrophobicity, not disulfide topology — correctly folded and misfolded isomers often co-elute. Reduced LC-MS chemically breaks all disulfide bonds (using DTT or TCEP) before analysis, reducing all cysteines to free sulfhydryl groups. This confirms the total number of cysteine residues but provides zero information about which cysteines are paired. Only non-reducing LC-MS combined with ETD/ECD fragmentation preserves the intact disulfide bond during ionisation and fragmentation, enabling direct assignment of cysteine pairings from the fragment ion mass differences.
What sample amount is required for disulfide mapping? +
Minimum 50–200 µg of purified peptide is recommended depending on the number of disulfide bonds and sequence length. Peptides with 2 disulfide bonds (e.g., α-conotoxins) require approximately 50 µg; 3-disulfide peptides (e.g., ω-conotoxins, cyclotides) require approximately 100 µg; complex knottins and miniproteins with 3+ disulfides require approximately 200 µg. Lower amounts may be discussed on a case-by-case basis for screening purposes — contact us with your available quantity before sample submission.
Can you analyse disulfide connectivity for peptides with more than three disulfide bonds? +
Yes. Peptides with four or more disulfide bonds are addressed using our extended partial reduction workflow, in which disulfide bonds are selectively and sequentially reduced under controlled TCEP exposure conditions. This generates overlapping connectivity subsets that collectively resolve the full cystine network. Interpretation complexity increases with disulfide count, and we will assess feasibility after reviewing the peptide sequence and available sample quantity before confirming the project scope.
Do you provide engineering recommendations after characterisation? +
Yes. For characterised hits, we offer a preliminary scientific consultation covering oxidative folding stability assessment, refolding protocol recommendations, and cyclisation strategy options to improve proteolytic resistance and developability. This is provided as expert scientific guidance — not as a guaranteed optimisation service — and is scoped in consultation with your project team before the study begins.
Can you analyse PTMs alongside disulfide connectivity in the same run? +
Yes. Our HRMS platform captures C-terminal amidation, N-terminal pyroglutamylation, hydroxylation, and O-/N-linked glycosylation modifications alongside disulfide connectivity in a single analytical run. PTM assignments are included in the structured report and are flagged as potentially affecting bioactivity where fragmentation quality permits site-specific localisation.
Case Study: Structural Identification of a Novel T1-Conotoxin with Non-Canonical Disulfide Connectivity
Journal: Journal of Venomous Animals and Toxins Including Tropical Diseases
Published: 2020
DOI: 10.1590/1678-9946202050502 (CC BY 4.0)
Research Context
Conotoxins are a structurally diverse family of disulfide-rich venom peptides produced by cone snails (Conus species), each targeting specific ion channels or receptors with high affinity and selectivity. The T1 conotoxin superfamily is characterised by a four-cysteine (CysI–CysIV) framework that, across the vast majority of characterised T1 members, adopts a conserved disulfide pairing pattern — typically CysI–CysIII and CysII–CysIV — that defines the geometry of the bioactive surface. This connectivity is the structural basis for receptor recognition, and even minor perturbations to the disulfide scaffold can abolish or dramatically alter pharmacological activity.
The study that yielded BN5A was driven by a broader screening effort to characterise the venom peptidome of Conus bandanus, a species known to produce pharmacologically active conotoxins targeting voltage-gated calcium and sodium channels. Unlike targeted toxin discovery programmes that begin with a known pharmacological target and screen for active fractions, this study adopted an unbiased peptidomics approach — fractionating crude venom by RP-HPLC, profiling each fraction by MALDI-TOF, and sequencing hits by tandem MS — with the aim of identifying novel sequences not predicted by transcriptomics.
Study Objectives
The primary objective was to isolate, sequence, and structurally characterise a novel T1-conotoxin from Conus bandanus venom. Specifically, the team sought to:
- Confirm the monoisotopic mass and cysteine content of the isolated peptide by MALDI-TOF
- Establish the linear amino acid sequence by reduction, alkylation, and CID-MS/MS
- Assign disulfide connectivity under native (non-reducing) conditions using CID-induced fragment ion pairs
- Compare the assigned connectivity against the canonical T1 framework to determine whether BN5A adopted the expected or an alternative pairing
Methods
Venom fractionation. Crude venom from Conus bandanus was extracted and fractionated by reversed-phase HPLC on a C18 column. Fractions were screened by MALDI-TOF; fractions containing peptide signals in the 1,000–5,000 Da range — consistent with conotoxin molecular weights — were retained for further analysis.
Intact mass and cysteine count. MALDI-TOF was used to determine the monoisotopic mass of the isolated peptide BN5A. The observed mass was consistent with a 14-residue peptide containing four reduced cysteine residues (Cys residues contribute 103.009 Da per sulfhydryl group in reduced form; in the oxidised (disulfide-linked) peptide, two cysteines lose 2.016 Da of hydrogen mass to form one S–S bond).
Linear sequence by reduced MS/MS. To establish the primary amino acid sequence, BN5A was fully reduced with TCEP to break all disulfide bonds, alkylated with iodoacetamide to derivatise the free sulfhydryl groups (preventing re-oxidation), and subjected to CID-MS/MS fragmentation. The resulting b- and y-ion series from CID sequencing established the 14-residue backbone sequence.
Disulfide connectivity by non-reducing CID-MS. The native (non-reduced, non-alkylated) peptide was then analysed by CID-MS/MS under conditions that fragment the peptide backbone but do not reduce the disulfide bonds. The mass differences between pairs of fragment ions linked by an intact disulfide bridge directly reveal which cysteine residues are paired. For BN5A, the diagnostic fragment ion pairs were identified and their masses matched to specific Cys pairings.
Key Findings
BN5A was confirmed as a 14-residue peptide with four cysteine residues at positions consistent with the T1 framework. MALDI-TOF intact mass matched the expected mass for a four-disulfide conotoxin (two disulfide bonds formed, one S–S bond per pair = net mass shift of 2.016 Da relative to the fully reduced form).
Reduced MS/MS sequencing established the linear backbone, confirming four cysteine positions. Critically, non-reducing CID-MS analysis identified a disulfide pairing of CysI–CysIV and CysII–CysIII — a pairing pattern that is the reverse of the canonical T1 connectivity (CysI–CysIII and CysII–CysIV). Figure 5 of Bao et al. (2020) shows the annotated CID spectrum with the key diagnostic fragment ion pairs used to confirm this non-canonical assignment.
The biological significance of this reversed pairing is substantial. In the canonical T1 fold, the CysI–CysIII bond constrains the N-terminal loop that typically contacts the receptor, while the CysII–CysIV bond positions the C-terminal region. The reversed connectivity in BN5A would reorient these structural elements, potentially altering which ion channel subtype the peptide targets and how it binds — making this a pharmacologically distinct conotoxin variant.
Implications for DRP Discovery Workflows
This case illustrates several principles directly relevant to any DRP characterisation programme:
- Canonical framework does not guarantee canonical connectivity. Even well-characterised conotoxin superfamilies can harbour members with unexpected disulfide pairings. Assuming connectivity based on framework type alone risks misinterpreting pharmacological data.
- Non-reducing MS is non-negotiable for connectivity assignment. The reversed CID-MS workflow — native peptide fragmentation without reduction — was the analytical step that resolved the BN5A connectivity. A reduced-only analysis would have reported only the total cysteine count and completely missed the non-canonical pairing.
- For more complex DRPs, ETD/ECD outperforms CID for connectivity assignment. BN5A has only four cysteine residues (two disulfide bonds), and CID fragmentation provided sufficient resolution. Peptides with three or more disulfide bonds — where fragment ion complexity increases combinatorially — benefit significantly from ETD/ECD fragmentation, which generates cleaner b- and y-ion series without the preferential cleavage and neutral loss patterns that complicate CID spectra.
- Folding homogeneity matters for pharmacology. The BN5A study focused on the correctly folded isomer, but synthetic or recombinant DRP production frequently yields a mixture of correctly folded, misfolded, and scrambled disulfide isomers. Oxidative folding assessment by non-reducing RP-HPLC — which separates isomers by hydrophobicity before MS confirmation — is essential for distinguishing the bioactive fraction from inactive contaminants.
How the Creative Proteomics Platform Addresses These Challenges
The BN5A characterisation workflow — MALDI-TOF intact mass → reduced CID sequencing → non-reducing CID connectivity assignment — maps directly onto the capabilities of the Creative Proteomics DRP platform. For four-Cys peptides, non-reducing CID-MS is appropriate; for more complex scaffolds (three or more disulfide bonds), ETD/ECD fragmentation and stepwise partial reduction provide the additional resolving power needed. Oxidative folding assessment by non-reducing RP-HPLC + HRMS adds the critical quality control step that distinguishes correctly folded from scrambled isomers — a step absent from both standard peptide synthesis QC and most general proteomics core workflows.
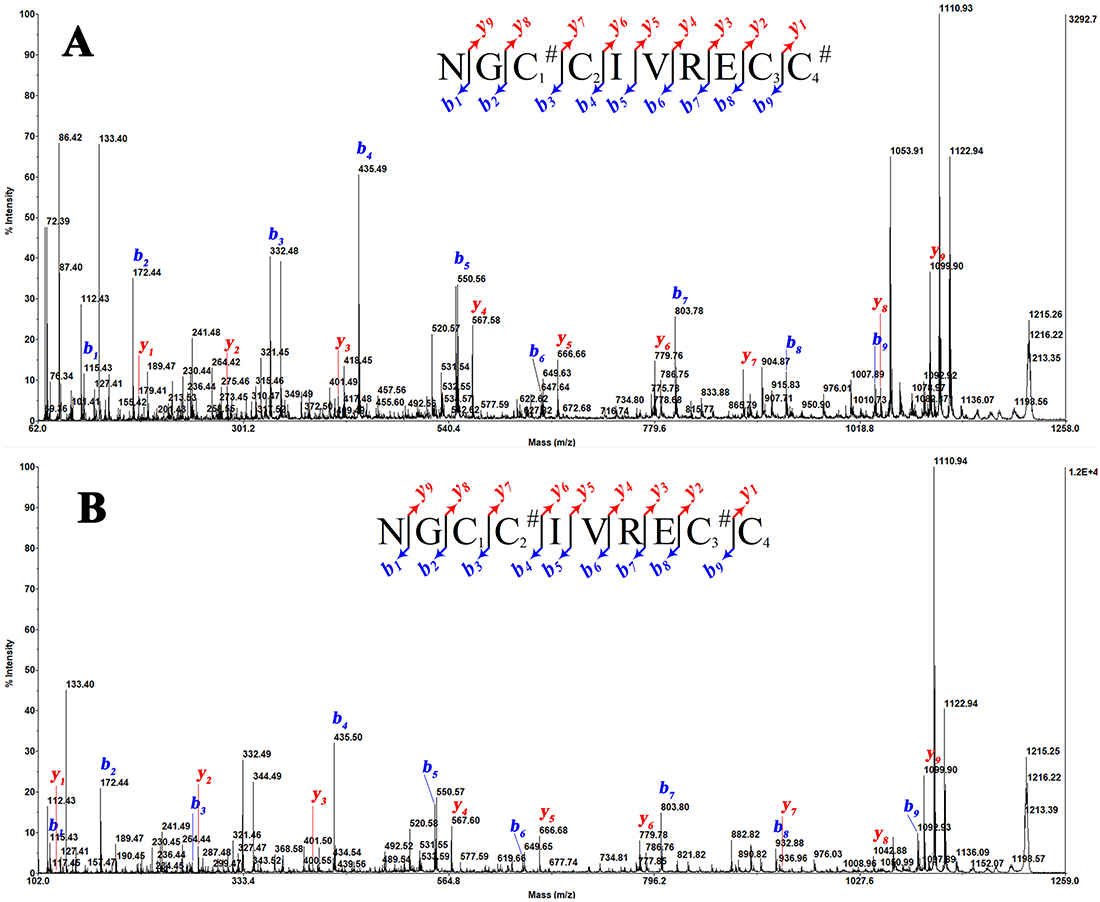 CID-MS/MS spectrum of the native BN5A conotoxin annotated with disulfide-linked fragment ion pairs. Non-canonical CysI–CysIV and CysII–CysIII connectivity confirmed. Inset: canonical (grey) vs reversed (orange) disulfide pairing. Source: Nguyen Bao et al., 2020, Journal of Venomous Animals and Toxins Including Tropical Diseases, Figure 5.
CID-MS/MS spectrum of the native BN5A conotoxin annotated with disulfide-linked fragment ion pairs. Non-canonical CysI–CysIV and CysII–CysIII connectivity confirmed. Inset: canonical (grey) vs reversed (orange) disulfide pairing. Source: Nguyen Bao et al., 2020, Journal of Venomous Animals and Toxins Including Tropical Diseases, Figure 5.