Published Research
Best Practices and Benchmarks for Intact Protein Analysis by Top-Down Mass Spectrometry
DOI
10.1038/s41592-019-0457-0
Challenge
Intact protein analysis by mass spectrometry — the foundation of top-down protein sequencing — presents unique challenges compared to peptide-level analysis. Large proteins such as monoclonal antibodies (~150 kDa) require specialized sample preparation to remove MS-incompatible buffer components without denaturing or losing the protein. Prior to this study led by the Consortium for Top-Down Proteomics, there were no standardized protocols or benchmarks for intact protein sample preparation and MS analysis, making it difficult for laboratories and CROs to establish reliable workflows.
Analytical Approach
- Three sample preparation protocols systematically evaluated across multiple laboratories and instrument platforms.
- Protocol 1 (Dilution) — tested for high-concentration protein samples with MS-compatible buffers.
- Protocol 2a (MWCO Filtration) — evaluated for buffer exchange applications requiring salt or detergent removal.
- Protocol 3 (Protein Precipitation) — assessed for detergent-containing samples such as those extracted with RIPA buffer.
- Intact protein mass spectra acquired on Orbitrap, FT-ICR, and QTOF instruments across nine laboratories, with standardized protein standards and NISTmAb reference material.
Relevance to Top-Down Protein Sequencing
- Demonstrates that intact monoclonal antibodies (NISTmAb, 150 kDa) can be characterized at the intact protein level with high mass accuracy.
- Establishes quantitative benchmarks for buffer compatibility — critical for successful sample preparation in any top-down sequencing project.
- Validates cross-platform consistency, confirming that results from different instrument types are comparable when standardized protocols are followed.
- Provides the foundation for the sample preparation protocols used in our top-down protein sequencing services.
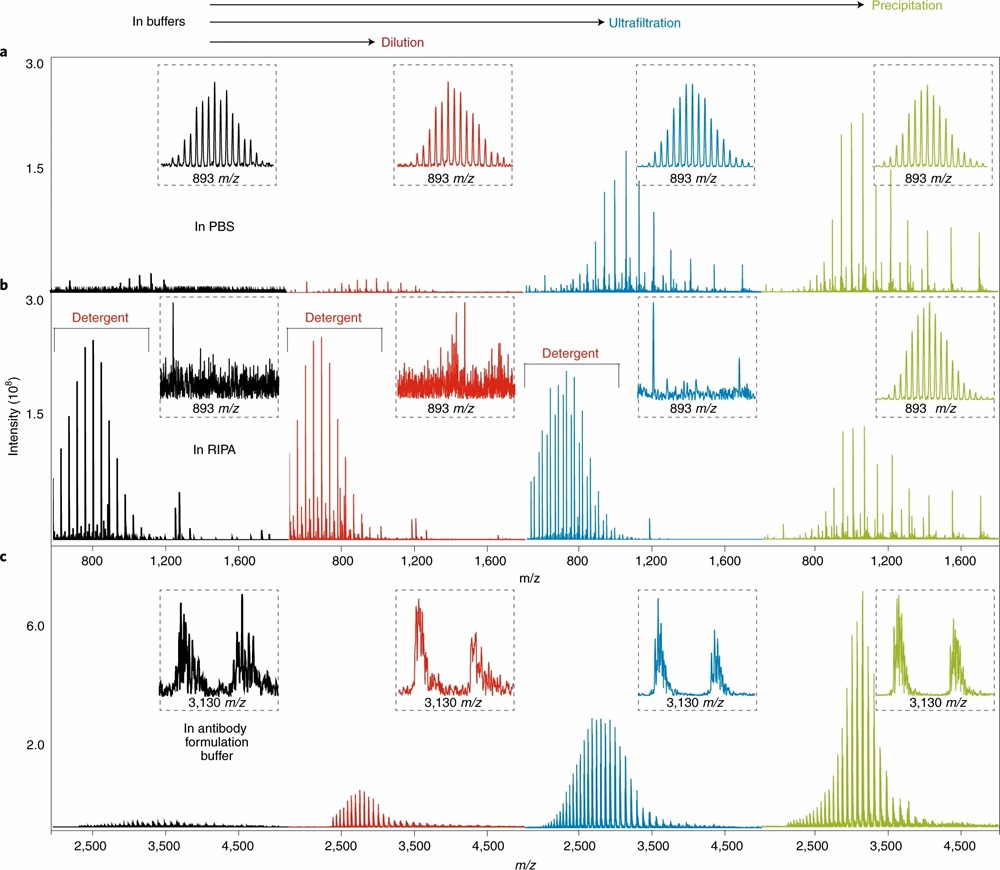
Intact mass analysis of NISTmAb and standard proteins demonstrating the benchmarks and best practices established for top-down mass spectrometry. (Adapted from Donnelly et al., 2019, Nature Methods)
Key Findings
Buffer Inhibition Thresholds
NaCl inhibits ESI-MS signal with SC₅₀ ≈ 1.5 mM; detergents produce the strongest signal suppression; volatile salts (ammonium acetate) are the most MS-compatible
Mass Accuracy Benchmarks
FT-MS instruments achieved ≤10 ppm mass accuracy; QTOF instruments achieved ≤20 ppm for intact protein measurements
Intact Antibody Success
NISTmAb (150 kDa) intact mass successfully measured after optimized sample preparation with clear glycoform resolution
Native vs Denaturing MS
Native MS produced approximately 2× the base peak intensity of denaturing MS for carbonic anhydrase at the same concentration
Publication Reference
Donnelly DP, Rawlins CM, DeHart CJ, et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat Methods. 2019;16(7):587–594. DOI: 10.1038/s41592-019-0457-0.