Protein Identification Services
- Home
- Applications
- Proteomics Analysis Services
- Protein Identification Services

Service Details
The identification and quantification of the proteins that a whole organism expresses under certain conditions is a main focus of proteomics, of which a main goal of proteomics is to identify proteins from complex mixtures. It is well known thatprotein identification is a matter of determining the right and wrong of a protein product. Protein molecules are very complex with many characteristic parameters. The most commonly used parameters, including protein molecular weight, protein isoelectric point, protein structure, protein biological function and immunological reaction, that can be measured at present. Interestingly, proteins are unique chains of variable length, made up of varying amino acids, so the obvious protein identification is to find its sequence of amino acids, of which the easiest ways to distinguish between proteins should be mass. Fortunately, it is possible for many different proteins to have nearly the different mass, and that mass spectrometry can help by giving almost exact masses.
Creative Proteomics offers the following featured protein identification services to assist your scientific research with years' experience in advanced experiment equipment.
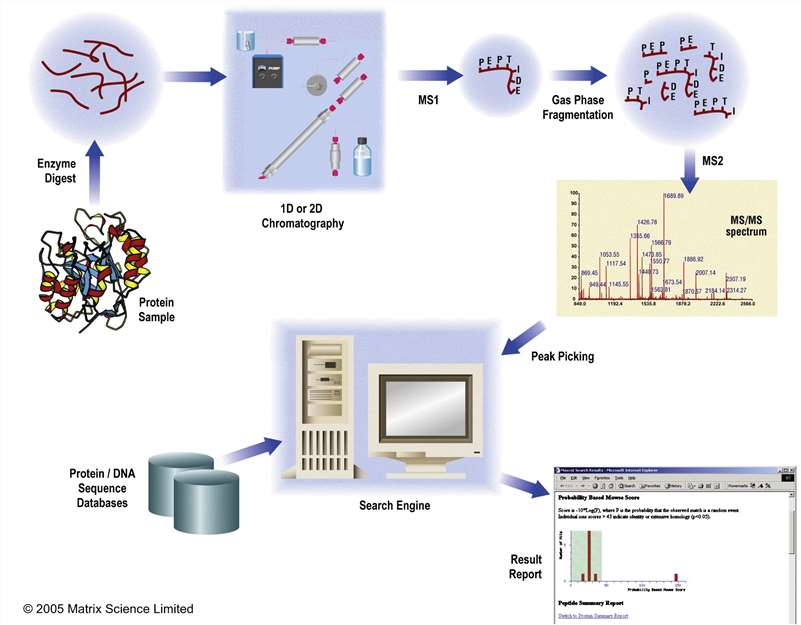 Fig. 1. A typical experimental workflow for protein identification and characterisation using MS/MS data. (Cottrell JS, 2011)
Fig. 1. A typical experimental workflow for protein identification and characterisation using MS/MS data. (Cottrell JS, 2011)
Two major strategies widely used for protein identification by mass spectrometry are MALDI-TOF-based protein fingerprinting and LC-MS/MS-based peptide sequencing. LC-MS/MS has higher sensitivity and accuracy than MALDI-TOF and can identify multiple protein components from a single sample.
Creative Proteomics uses LC-MS/MS technology to identify proteins in various samples, including gel samples (SDS-PAGE, native PAGE), IP eluates, body fluids, and tissues.
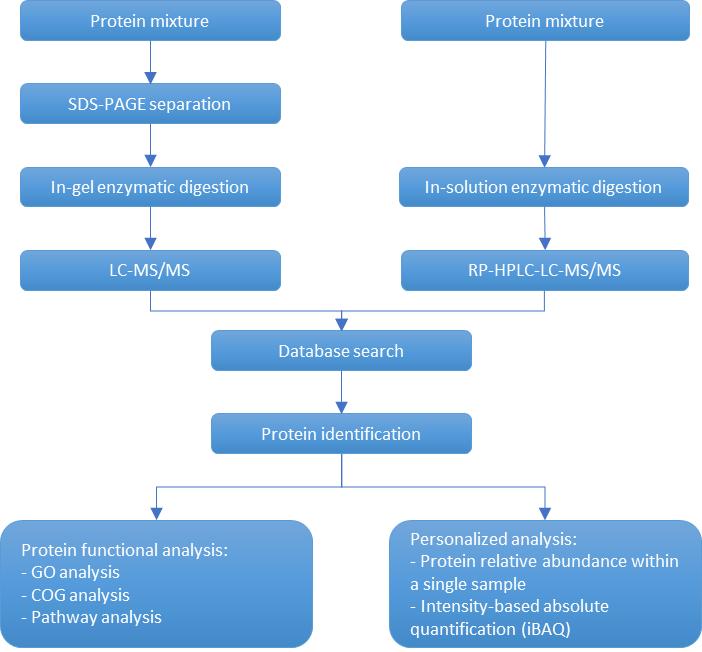 Process of protein gel band/mixed solution identification
Process of protein gel band/mixed solution identification
Technical Features
Scope of Application: Species with available protein databases or known target protein sequences
For samples that cannot be identified by available database analysis, de novo protein sequence analysis can be used.
Protein synthesis typically starts from the N-terminus, making the sequential composition of this end crucial for the protein's overall biological function. Analyzing the N-terminus through sequencing can reveal important information about the protein's complex structure and biological functions.
Additionally, the C-terminus serves as a significant structural and functional site for the protein. Post-translational modifications, such as C-terminal cleavage, can impact protein structure and biological function. Therefore, identifying the amino acid compositions and sequences of the C-terminus can aid in revealing the protein's structure and function.
Creative Proteomic utilized advanced LC-MS/MS technology, combined with labeling methods, to identify protein N/C-termini with high confidence.
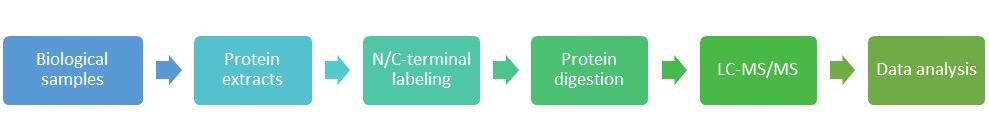 Process of protein N/C-termini identification based on LC/MS-MS
Process of protein N/C-termini identification based on LC/MS-MS
MALDI-ISD is a top-down proteomics approach that cleaves inter-residue bonds of the target protein, mainly the N-Cα bond of the main chain, to obtain molecular and fragment ion mass data through ionization and dissociation in a mass spectrometer. The mass difference between consecutive fragment ions is used to identify the N/C-terminal sequence of the target protein.
 Process of protein N/C-termini identification based on MALDI-ISD
Process of protein N/C-termini identification based on MALDI-ISD
Technical Features
Scope of Application: Suitable for all types of N/C-terminal sequence detection
| Sample Type/Product Type | Gel Spot Identification | Gel Stripe Identification |
|---|---|---|
| Stained/Silver-stained Gel Spot/Stripe | Visible spots | - |
| Protein Amount (per spot/stripe) | ≥1μg | ≥5μg, concentration ≥0.5μg/μL |
| Single Protein (e.g., for IP, Co-IP purified protein | - | - |
Notes: Gel silver staining should not contain glutaraldehyde; Preferably submit stained samples as false positives may occur with silver staining; Ensure gel cutting and submission within one-week post-electrophoresis.
| Sample Type | Submission Amount | Remarks |
|---|---|---|
| Fresh Animal Tissue (dry weight) | ≥10mg | - |
| Plant/Fungal Samples (e.g., mushrooms, fungi) (wet weight) | ≥500mg | For seeds and fruits, which contain higher starch and sugar content, to avoid insufficient protein extraction and subsequent resubmission, submit at least 2g. |
| Microorganisms (e.g., yeast, molds, bacteria, bacteriophages) | ≥50mg or ≥5×106 cells | - |
| Freshly Cultured Cells (number of cells) | ≥1×107 | - |
| Isolated Exosomes provided by the customer | ≥100μg, concentration ≥0.5μg/µL | - |
| Blood Samples (depleted of high abundance proteins) | Serum, Plasma ≥200µl | Whole blood is generally not recommended due to cell lysis upon thawing, affecting protein analysis. |
| Sample Type | Submission Amount |
|---|---|
| Protein Liquid | ≥200μg, concentration ≥0.5µg/µL |
 Biological Research: Unveiling protein functions and interactions in cells and organisms, elucidating biological processes.
Biological Research: Unveiling protein functions and interactions in cells and organisms, elucidating biological processes.
 Disease Diagnosis and Biomarker Discovery: Identifying disease-associated protein patterns for early diagnosis and personalized treatment strategies.
Disease Diagnosis and Biomarker Discovery: Identifying disease-associated protein patterns for early diagnosis and personalized treatment strategies.
 Drug Discovery and Development: Identifying drug targets and evaluating candidate compounds for novel therapeutics with improved efficacy.
Drug Discovery and Development: Identifying drug targets and evaluating candidate compounds for novel therapeutics with improved efficacy.
 Proteomics: Large-scale study of proteins, including expression patterns, post-translational modifications, and interactions.
Proteomics: Large-scale study of proteins, including expression patterns, post-translational modifications, and interactions.
 Food and Agriculture: Ensuring food safety, quality control, and nutritional assessment through protein analysis.
Food and Agriculture: Ensuring food safety, quality control, and nutritional assessment through protein analysis.
 Environmental Monitoring: Assessing the impact of pollutants on organisms and ecosystems through protein expression analysis.
Environmental Monitoring: Assessing the impact of pollutants on organisms and ecosystems through protein expression analysis.
Creative Proteomics is a trusted provider of proteomics analysis services. Thanks to our outstanding scientists and powerful mass spectrometry platform, we offer a range of protein identification services to our clients. Our service guarantees accurate and reliable results, at quick turnaround time! If you are interested in our services, please contact us to see how we can help solve your problem.
References
1. How many samples are needed for protein identification using MALDI-TOF/TOF?
For spot or band samples from 2D gels, as long as they are visible to the naked eye, a relatively high success rate can be ensured. Alternatively, providing greater than 200 fmol (or approximately 10 ng of a protein around 50 kDa) also yields a high success rate. For solution or freeze-dried samples, a protein content of at least 100 ng is required.
2. When performing protein spot identification using 2-DE with silver staining, which silver staining reagents might affect subsequent mass spectrometry identification?
Avoid the use of glutaraldehyde in silver staining reagents, as it can modify proteins and thus affect subsequent protein identification. Refer to silver staining methods compatible with mass spectrometry for this staining method.
3. How to reduce keratin contamination?
Keratin contamination mainly arises from hair and skin flakes, so it is advisable to wear gloves and a head covering during experiments and gel cutting.
4. Is it necessary to merge gel spots?
For stained samples, as long as a spot is visible to the naked eye, it is sufficient. For silver-stained samples, it is recommended to merge four individual spots, but in general, we suggest using LC-MS/MS for silver-stained sample identification.
5. Regarding sample storage time?
We recommend analyzing freshly prepared samples immediately. The storage time for silver-stained samples should ideally not exceed 2 months, while stained samples can be stored for several months, but preferably not more than 6 months.
6. Do you accept enzymatically digested samples?
Apart from trypsin, are other enzymes used for digestion? We do not accept pre-digested samples as we have strict QC controls for digestion that require our intervention. Typically, we use trypsin alone for digestion. If other enzymes are needed for digestion, please contact us in advance.
7. How to identify low molecular weight proteins?
Generally, if the molecular weight is above 20 kDa, we recommend MALDI-TOF/TOF for identification; if it is below 20 kDa, we recommend LC-MS/MS.
8. Sample shipping arrangements?
For gel spots or bands, place them directly into 1.5 mL EP tubes (preferably imported; if not available, please contact us, and we will provide imported EP tubes free of charge). Write the sample number on the tube lid. No liquid needs to be added to the tube. Fill out the sample submission form and send it at room temperature. For other purified or freeze-dried samples, if stable at room temperature, they can be sent at room temperature; otherwise, dry ice shipment is recommended.
9. When comparing identified proteins to the bidirectional protein map, discrepancies arise, such as differing molecular weights or isoelectric points. In these cases, how can one ascertain the most credible protein among several or even dozens identified within a spot?
Protein identification by mass spectrometry involves mapping peptides to proteins. Therefore, some identified proteins may be homologous proteins of the target protein or subunits of the protein. Additionally, protein modifications or degradation can affect the protein's position on the gel. Generally, the higher the Mascot identification score, the higher the credibility. Furthermore, factors such as molecular weight, isoelectric point, peptide coverage, etc., can be considered to select proteins with higher credibility.
Vibrio parahaemolyticus is a significant pathogen in aquatic animals, causing severe economic losses in aquaculture. Understanding the immune response of aquatic organisms to V. parahaemolyticus infection is crucial for disease management and prevention. Tetraodon nigroviridis, a model organism in aquatic research, provides valuable insights into host-pathogen interactions.
Spleen samples from T. nigroviridis infected with V. parahaemolyticus were collected for transcriptomic, proteomic, and miRNA analyses. These samples provided insights into the molecular mechanisms underlying the host-pathogen interaction.
Transcriptome Sequencing: Total RNA was extracted from spleen samples of T. nigroviridis infected with V. parahaemolyticus and control samples. RNA integrity was assessed, and libraries were prepared using the Illumina TruSeq Stranded mRNA Library Prep Kit. Paired-end sequencing was performed on an Illumina platform to generate high-quality reads. Raw sequencing data were filtered to remove adapters and low-quality reads. The clean reads were aligned to the T. nigroviridis reference genome using software such as HISAT2 or STAR. Differential expression analysis was conducted using tools like DESeq2 or edgeR, with DEGs identified based on fold change and statistical significance.
Proteome Analysis: Protein extraction was performed from spleen samples using methods such as trichloroacetic acid (TCA)/acetone precipitation or protein lysis buffer. Protein concentration was quantified using the Bradford or BCA assay. Proteins were digested into peptides using trypsin, and peptide mixtures were analyzed by LC-MS/MS. Data analysis involved peptide identification using database search algorithms like Mascot or Sequest, followed by protein quantification and annotation. DEPs were determined based on abundance differences between infected and control samples.
miRNA Profiling: Small RNA was isolated from spleen samples using techniques such as phenol-chloroform extraction or commercial kits. Libraries for small RNA sequencing were constructed using the NEBNext Multiplex Small RNA Library Prep Set for Illumina. Sequencing was performed on an Illumina platform to obtain raw miRNA sequencing data. Quality control steps were applied to remove adapter sequences and low-quality reads. Clean miRNA reads were aligned to miRBase or the T. nigroviridis genome to identify known and novel miRNAs. Differential expression analysis was conducted using tools like DESeq2 or edgeR to identify DE miRNAs between infected and control groups.
High Pathogenicity of V. Parahaemolyticus: Infected T. nigroviridis exhibited mortality, pathological symptoms, and tissue damage, highlighting the virulence of V. parahaemolyticus.
Identification of Immune Factors: Transcriptomic and proteomic analyses revealed differential expression of immune-related genes and proteins, including chemokines, interleukins, interferons, and complement factors.
Complement System Activation: Integrated analysis identified C3 and C1qA as key components of the complement system involved in the host response to V. parahaemolyticus infection.
Evolutionary Patterns of Immune Genes: Phylogenetic analysis showed variations in the number of C3 genes among fish species, indicating adaptation to diverse environmental and pathogenic pressures.
Theoretical Model of Host Response: A theoretical model integrating multi-omics data was proposed, elucidating immune response mechanisms and key regulatory pathways in T. nigroviridis infected with V. parahaemolyticus.
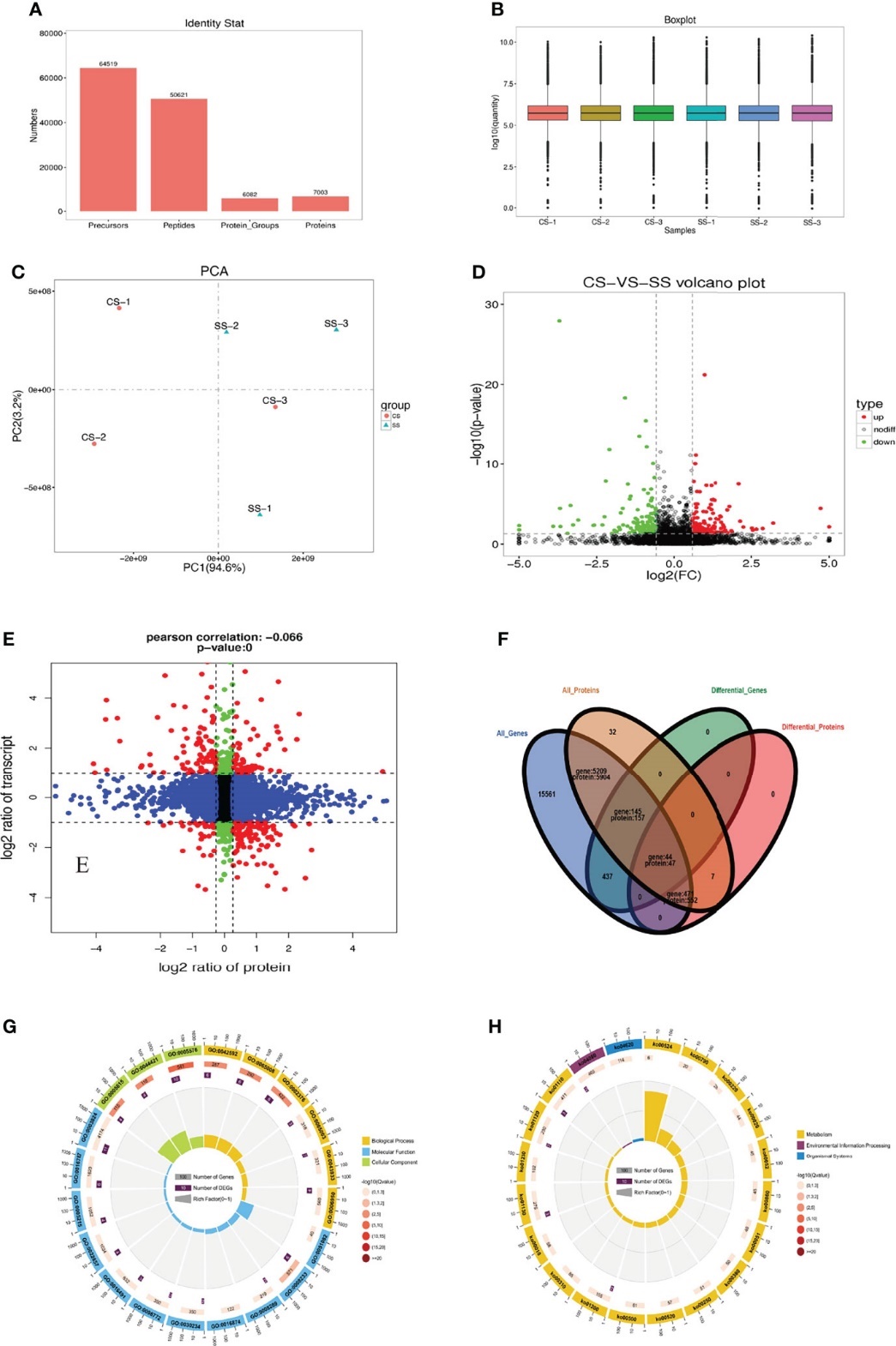 Statistics of protein identification by DIA
Statistics of protein identification by DIA
Reference
For research use only, not intended for any clinical use.