Published Research Example
XA-Novo: High-Throughput MS-Based De Novo Sequencing for Monoclonal Antibodies
Journal
Nature Communications
Project Overview
Researchers at Xiamen University developed an integrated platform combining single-pot multi-enzymatic gradient digestion (SP-MEGD) with deep learning-based de novo peptide sequencing. The method was validated on six COVID-19 neutralizing antibodies with known sequences (S2P6, SA55, SA58 human; 85F7, 36H6, 2B4 murine) and applied to six commercial therapeutic antibodies with unknown sequences, consistently achieving 100% sequence coverage across heavy and light chains.
Analytical Approach
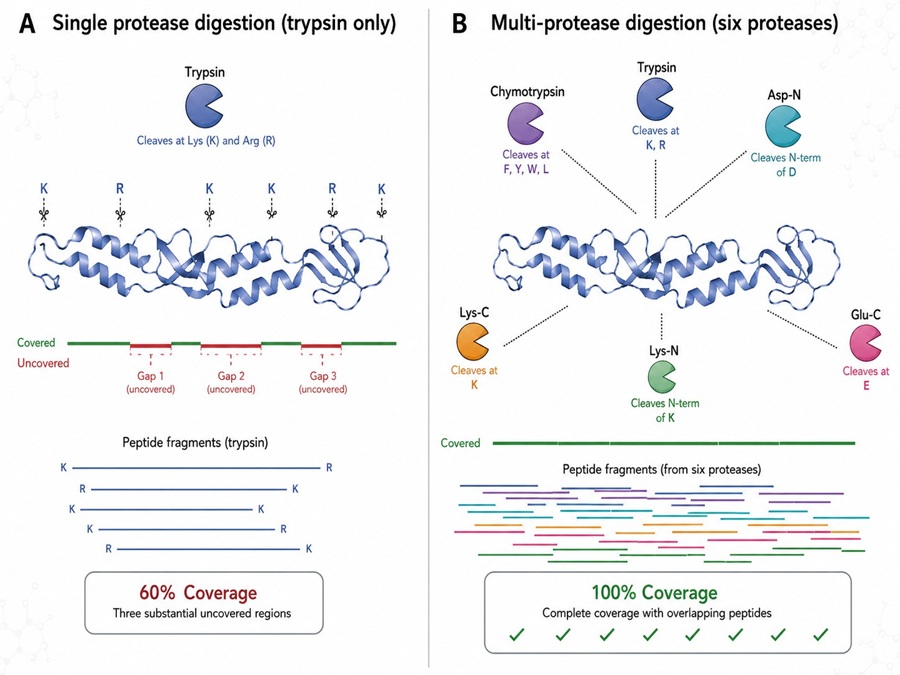
- SP-MEGD digestion with five proteases — trypsin, chymotrypsin, pepsin, elastase, and Asp-N — in a single-pot timed gradient with fractions at 2, 4, and 6 hours pooled for maximum peptide diversity.
- LC-MS/MS on Thermo Scientific Orbitrap Eclipse with dual fragmentation (HCD stepped collision energy for backbone sequencing, EThcD for regions lacking basic residues).
- Deep learning de novo sequencing via the Casanovo transformer model, followed by beam search assembly (beam size = 5) reconstructing full-length antibody sequences.
Relevance to Full-Length Sequencing
- Demonstrates that multi-protease digestion achieves 100% sequence coverage — the same core strategy deployed in our full-length sequencing service.
- Single amino acid accuracy of 100% in CDR regions, proving multi-protease approaches can discriminate even the most challenging sequence variants including Leu/Ile.
- The SP-MEGD approach parallels our continuous digestion method, confirming multi-enzyme strategies as the definitive solution to the sequence coverage problem.
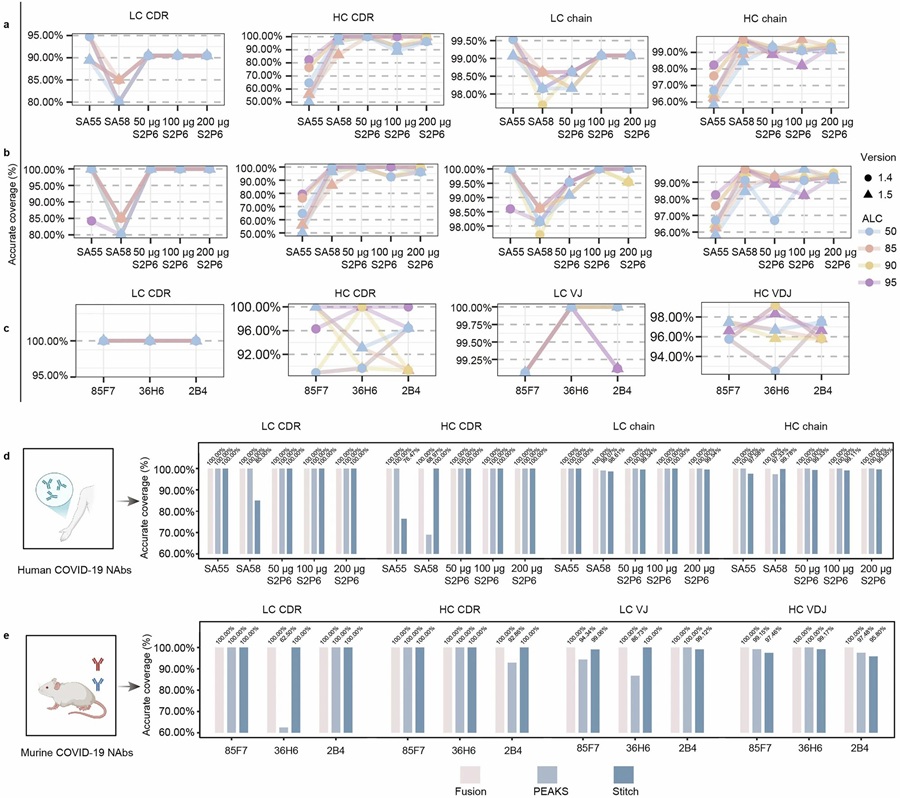
Sequence coverage comparison across antibodies demonstrating 100% coverage for all tested samples using the multi-protease gradient digestion approach. (Adapted from Xiong et al., 2026, Nature Communications)
Key Findings
100% Sequence Coverage
Heavy and light chains fully covered for all six tested antibodies
100% CDR Accuracy
All six CDR regions sequenced with perfect accuracy; Leu/Ile resolved
3-Antibody Mixtures
Simultaneous full-length sequencing from a single sample at ≥99.54% accuracy
50 μg Input
Low sample requirement — practical for early-stage development
Publication Reference
Xiong Y, Jiang W, Xiao J, et al. XA-Novo: high-throughput mass spectrometry-based de novo sequencing technology for monoclonal antibodies and antibody mixtures. Nat Commun. 2026;17:3391. DOI: 10.1038/s41467-026-70496-y.