psychology
Expert Data Review
Automated search engines flag peptide-spectrum matches, but commercial software is not infallible. Our mass spectrometrists manually inspect each assignment — catching false positives that would otherwise appear in your report. This is the quality difference between a software-generated output and a scientist-validated result.
auto_awesome
Adaptive Protease Strategy
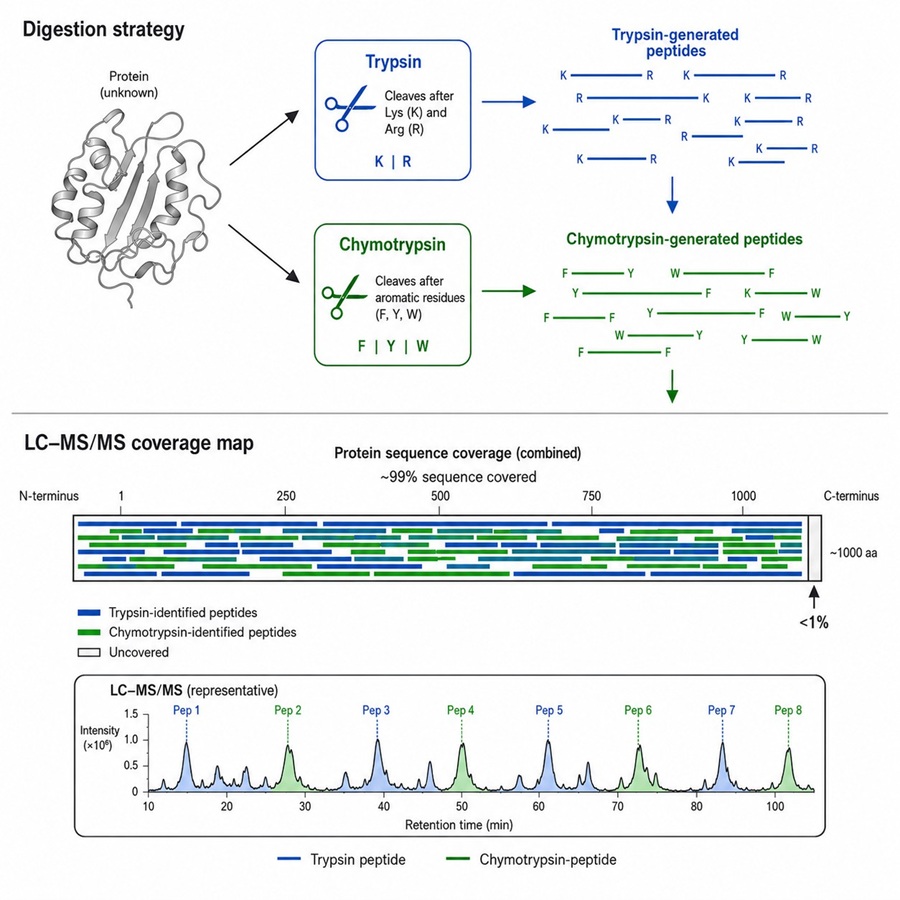
We do not default to trypsin-only digestion. Each project begins with an in silico analysis of your protein sequence, followed by selection of the optimal protease or protease pair — ensuring coverage that meets regulatory expectations out of the box.
description
ICH Q6B-Ready Reporting
Deliverables are structured for direct inclusion in regulatory filings. Sequence coverage maps, peptide identification tables, annotated MS/MS spectra, and PTM quantification summaries — organized to answer the questions reviewers will ask.