What Is Protein Characterization
Proteins are polymers formed by various amino acids and function biologically as protein drugs only when correctly folded into a specific conformation. Specific positions in the amino acid sequence can covalently bind with chemical groups, undergoing post-translational modifications. These modifications lead to structural changes in the protein, thereby affecting the biological activity of protein drugs. Consequently, it becomes necessary to assess the protein's molecular weight, coverage of peptide segments, and post-translational modifications.
The analysis of protein characterization aims to delineate the biological functions and diverse properties and parameters of proteins. This encompasses the assessment of protein type, content, molecular mass, amino acid composition, primary structure, and purity, among other factors. Additionally, the characterization extends to delineating the types of post-translational modifications, their respective sites, content, and interactions with other proteins. The predominant and advanced methodology employed in protein characterization is mass spectrometry-based technology. The general workflow of this technique involves the enzymatic digestion of proteins, followed by high-performance liquid chromatography (HPLC) for peptide separation, peptide mass spectrometry (MS) for detection, and subsequent generation and analysis of mass spectrometry data.
Physicochemical analysis of protein drugs
Molecular Weight Characterization
The measurement of molecular weight is a critical first step in identifying proteins. Employing high-resolution mass spectrometry allows us to obtain a multi-charge signal specific to proteins. The deconvolution of this signal provides an accurate molecular weight value and facilitates an preliminary evaluation of the protein's modification status. For antibody drugs, further analysis can be conducted by disassembling the light and heavy chains or by removing glycans, thus enabling specific evaluation of the molecular weight of both glycosylated and non-glycosylated light and heavy chains.
When analyzing polypeptide protein-based medications, molecular weight characterization provides a precise measure of their molecular weight. Specific proteins evaluated commonly include antibodies (measurable as a complete structure, with glycans removed, or in reduced form) and growth factors. Polypeptides assessed typically comprise insulin, carbocyclic oxytocin, and terlipressin among others. By supplying a theoretical sequence, we can accurately align it with its modification status, providing a more detailed analysis.
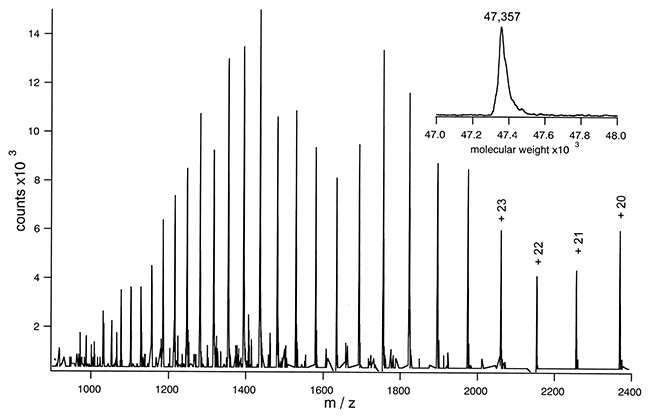 ESI Mass Spectrum of Lac Permease
ESI Mass Spectrum of Lac PermeaseHPLC Purity Analysis
Purity assessment and peptide mapping of protein peptides involve the utilization of high-performance liquid chromatography (HPLC). This technique facilitates the determination of protein purity and peptide distribution subsequent to enzymatic digestion, holding substantial importance for advancing research in protein peptide drugs. Purity analysis serves to scrutinize impurities and content within industrial protein peptide samples. Concurrently, peptide profile analysis is employed for the identification of peptide distribution, enabling the observation of consistency across various batches of test materials.
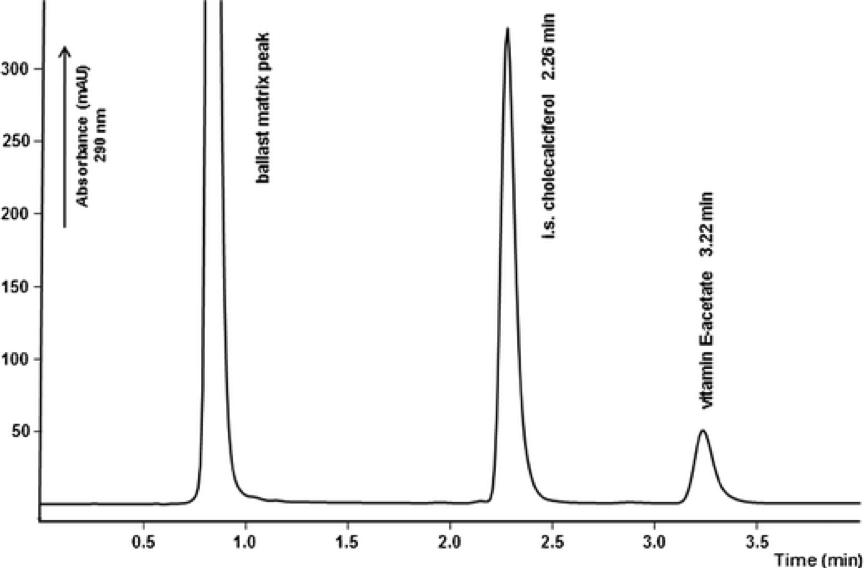 Fast HPLC method
Fast HPLC methodProtein Charge Variants Analysis
Charge variants analysis is essential in characterizing the efficacy and stability of protein therapeutics, especially monoclonal antibodies. Variation in protein charge, resulting from post-translation modifications, can impact a protein's physicochemical properties and biological activity. Therefore, it is critical to perform a comprehensive analysis of charge variants to ensure the quality and consistency of therapeutic proteins. Methods such as ion-exchange chromatography (IEX), hydrophobic interaction chromatography (HIC), and capillary electrophoresis (CE) are often used, supplemented with mass spectrometry for variant identification.
Select Service
Aggregation and Fragment Content Analysis
Structural fluctuations or local conformational changes in proteins might expose aggregation-prone sequences or "hot spots", subsequently leading to the formation of dimers and/or oligomers. Throughout the development process of biological drugs and the entire production phase, it's mandatory to identify and monitor charge isoforms. With the use of Size Exclusion Chromatography High-Performance Liquid Chromatography (SEC-HPLC), one can separate and analyze the content of monomer and polymer molecules.
Select Service
Identification and Quantification of Host Cell Protein Residues
The U.S. Pharmacopoeia has explicitly stipulated that LC-MS/MS technology should be utilized to identify and quantify host cell protein residues in biopharmaceuticals and establish quality standards for the control of such impurities. Analyses of host cell protein residues should be carried out in various batches of raw materials and end products. Given the low concentration of host cell protein residues, their analysis can be influenced by the main components of the drug. Therefore, careful consideration is required when choosing technical methods to identify and quantify them accurately and comprehensively.
Structural Characterization of Protein Drugs
Sequence Coverage/Peptide Mapping
Peptide mapping involves enzymatic digestion of proteins (typically using trypsin) to generate peptide fragments, which are then subjected to reproducible separation and identification. This process allows the detection and monitoring of minor changes as single amino acid variations, oxidation, deamidation, and other degradation products. Peptide map analysis can also directly detect common monoclonal antibody variants, such as N-terminal cyclization, C-terminal lysine loss, and N-glycosylation, as well as other post-translational modifications.
A peptide map, also known as a "protein fingerprint" represents the final product of a protein that has been subjected to enzymatic digestion and chromatographic separation. This comprehensive and in-depth characterization tool includes four main stages: protein isolation and purification; selective cleavage of peptide bonds; chromatographic separation of peptides; and confirmatory analysis of peptides. Peptide maps can determine primary protein structure and detect structural changes. Furthermore, they can confirm process consistency and genetic stability. Ideally, a peptide map should include positive protein identification and maximum coverage of intact peptide sequences.
Applications of peptide map mass spectrometry include:
Detailed protein characterization, such as the confirmation of N-terminal and C-terminal amino acid sequences, peptide coverage, and full polypeptide sequence.
Screening and identification of post-translational modifications, such as glycosylation, disulfide bonds, oxidation, glycation, phosphorylation, deamidation, N-terminal cyclization, and C-terminal lysine loss.
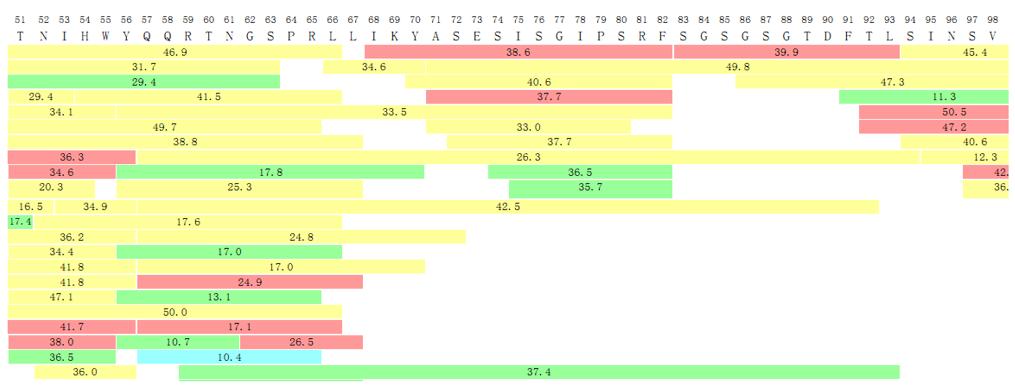
N-terminal and C-terminal Sequence Confirmation
Antibodies or recombinant proteins exhibit susceptibility to structural variations, including instances of incomplete signal peptide sequence processing at the N-terminus, variations in methionine residues, and modifications through glutamic acid cyclization. Truncation frequently occurs at the C-terminus, exemplified by the absence of a lysine residue in the C-terminus of the heavy chain of a monoclonal antibody. Consequently, a comprehensive analysis of the terminal amino acid sequences of the product becomes imperative for elucidating the characteristics and uniformity pertaining to both the amino and carboxyl termini of the protein.
Post-Translational Modification Analysis
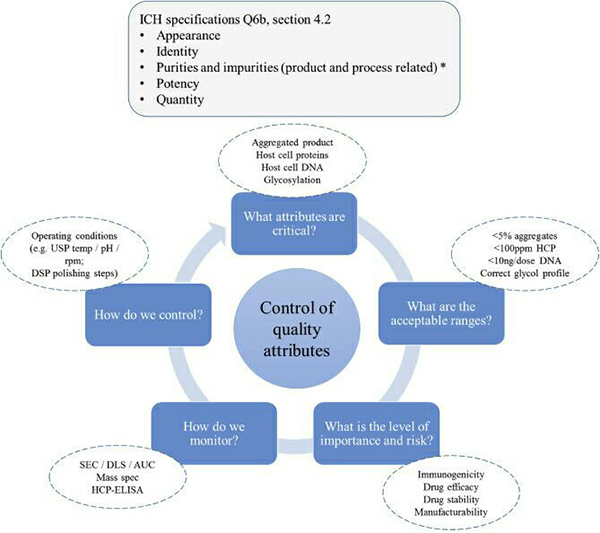
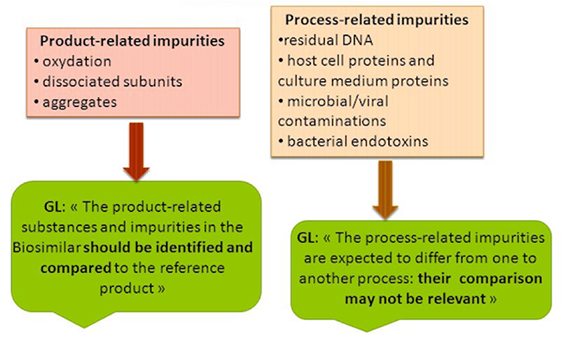
Post-translational modifications are ubiquitous in various recombinant proteins and antibodies, which lead to product heterogeneity and impact the quality. The ICH-Q6B guidelines stipulate that the forms of modifications such as deamidation, phosphorylation, glycosylation, and oxidation in biopharmaceuticals should be analyzed to establish quality acceptance standards. High-accuracy mass spectrometry combined with professional analytical software can be used to analyze the types of post-translational modifications present on the protein and their proportions.
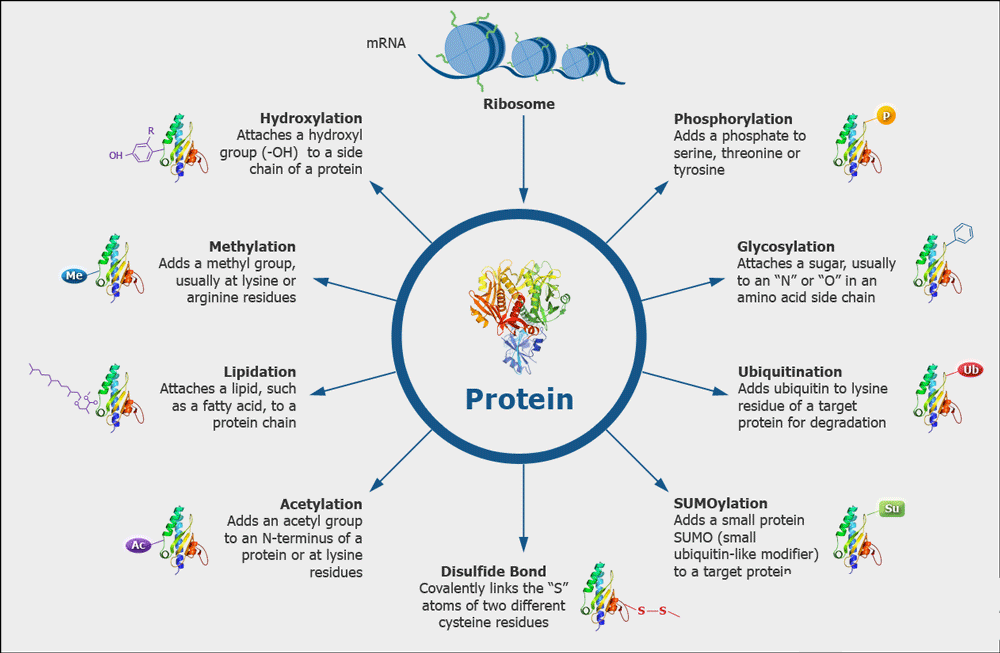
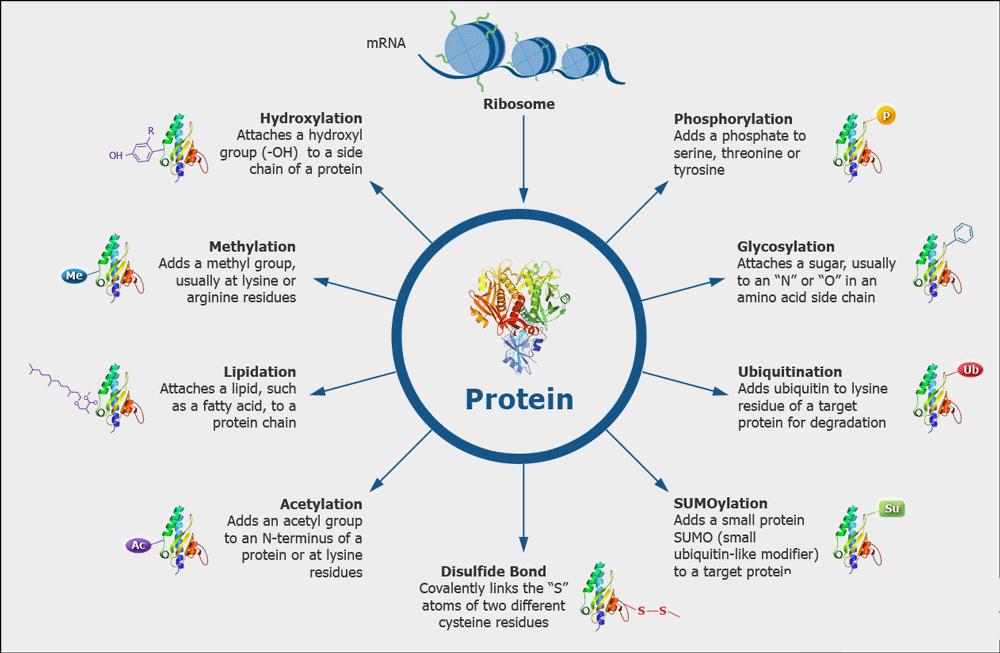 Various Kinds of Protein Modifications
Various Kinds of Protein ModificationsDisulfide Bond Analysis
An analysis of disulfide bonds can be utilized in the structural analysis of biotherapeutics, aiding in molecular design, property optimization and process studies. By parsing peptide spectrum data under non-reducing conditions using specialised software, critical disulfide bond location information can be gleaned.
Select Service
- Phosphorylation Analysis
- Deamidation and Oxidation Analysis
- Disulfide Bridges & Free Sulfhydryl Groups
- N-acetylation Analysis
- Methylation Analysis
- Ubiquitination Analysis
- Sumoylation Analysis
- Lipidation Analysis
- S-nitrosylation Analysis
- N-myristoylation Analysis
- S-palmitoylation Analysis
- S-prenylation Analysis
Glycosylation Modification Analysis
Glycosylation modification, unlike other modifications, has an extremely high heterogeneity and is an important factor contributing to protein structural diversity. The structure of the glycan plays a crucial role in complement activation and receptor affinity, making glycosylation modification a key attribute that can impact the efficacy of therapeutic antibodies. Additionally, the glycosylation types from non-human sources may trigger immune responses, leading to safety issues. Therefore, glycosylation modification analysis is an important aspect of characterizing therapeutic glycoproteins, particularly monoclonal antibodies. Guidelines from ICH-Q6B stipulate that the glycosylation sites, glycan structure (glycoform), and content of glycoproteins should be clearly identified. The complexity of glycosylation modification demands high-level analytical techniques. Only with an effective enrichment approach, high-precision LC-MS/MS, and professional data analysis software, can glycosylation be effectively characterized.
Analysis of Glycosylation Sites and Glycoforms
During drug screening phases, site-specific glycosylation, types of glycan, and the relative abundance of glycoforms can be simultaneously analyzed via glycopeptide profiling.
Oligosaccharide Spectrum Analysis
Oligosaccharide Spectrum Analysis involves exploiting pNGaseF enzyme to enzymatically cleave sugars from glycoproteins, and marking these cleaved sugar chains derivatively with a 2-AB reagent. High precision mass spectrometry is then employed to identify the types of sugar chains, and fluorescence signals are used for quantitative analysis of sugar chains. In the clearance phase, fluorescence chromatography alone can be used for quantitative analysis of the sugar spectrum.
Analysis of Sialic Acid
Sialic acid plays a crucial role in maintaining the stability and spatial conformation of glycoprotein. The sialic acid on the N-glycan and O-glycan of antibody drugs mainly consists of N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). Since the human body cannot synthesize Neu5Gc, it carries some immunogenicity. To ensure the safety and efficacy of drugs and to maintain the consistency of quality control between batches, ICH Q6B stipulates that the structure and relative content of sialic acid must be monitored during process development, manufacturing purification, formulation development, and stability testing. During the detection, the sialic acid on the protein is first hydrolyzed, and then the sialic acid content is accurately quantified based on the standard curve using triple quadrupole LC-MS/MS (QTRAP4500).
Select Service
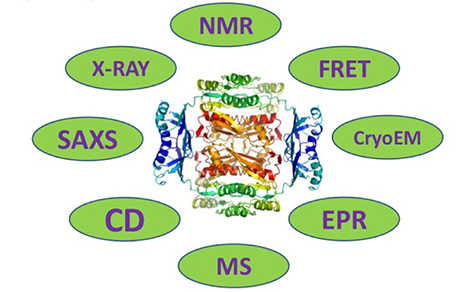
Protein Characterization Technology
Mass Spectrometry Characterization
Mass spectrometry uses the de novo sequencing method, where the amino acid sequence is inferred based on the mass difference between a series of orderly fragment ions produced by the collision between peptide and inert gas. We can deduce the amino acid sequence by focusing on the y ions and b ions at the peptide bond fracture site, thereby not being limited by post-translational modifications and purity. However, there are limitations to mass spectrometry sequencing, which includes: 1. Dependence on public databases- if the genomes of the studied species have not been completely sequenced, or if there are no corresponding sequences, then it will be impossible to correctly identify through mass spectrometry. 2. The scoring and algorithms of the search engine in use may overlook some peptide and mass spectra matches, thereby yielding false-positive results. 3. In instances of point mutations or unknown modifications, due to not being considered in the search algorithm or parameter settings, accurate results cannot be obtained. With the rapid advancements in more powerful instruments, analytical software, and databases, the advantages of mass spectrometry in the field of protein de novo sequencing will become increasingly apparent.
Mass spectrometry techniques are also widely used for protein purity identification, molecular mass determination, peptide (mass) spectrum analysis, disulfide bonds, acetylation, glycosylation, phosphorylation, and noncovalent binding, among others.
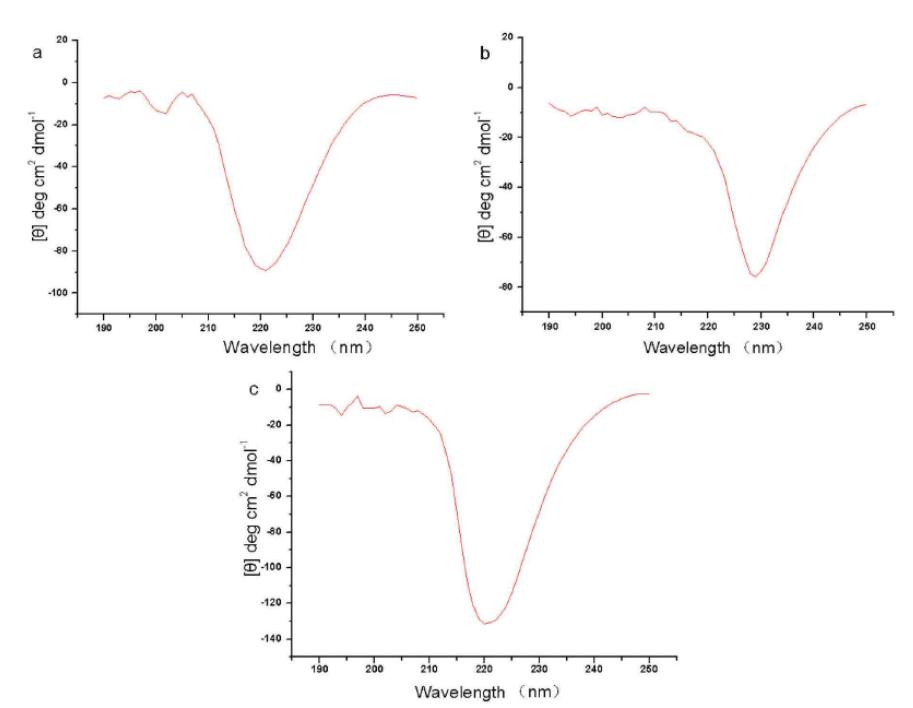
Characterization by Circular Dichroism Spectroscopy
Circular Dichroism (CD) Spectroscopy measures the dichroism of large biological molecules, thereby obtaining the secondary structure of these molecules. When plane-polarized light passes through a medium with optical activity, circular dichroism is produced due to the unequal absorption of the resulting right and left circularly polarized light by different chiral conformations of the same optically active molecule present in the medium.
The far-ultraviolet CD spectrum can be used to determine the secondary structure of proteins, while the near-ultraviolet CD spectrum can be used to assess the tertiary structure of protein side chains.
Select Service
Dynamic Light Scattering (DLS) Analysis
DLS is a common method used to determine the size distribution of small particles in suspension or polymers in solution. In this method, the particles are illuminated by a laser, and the fluctuation of light scattering intensity due to Brownian motion is measured. The autocorrelation function of the light scattering intensity fluctuation is calculated, and the particle size distribution is obtained by Laplace transform or non-negative least square method from the correlation function. In the biopharmaceutical industry, DLS has been widely used for the characterization of protein aggregation, formulation and stability studies, and sub-visible particle detection. As a non-invasive and rapid detection technique, DLS can provide the average size, size distribution, and related stability parameters of particles, and has the characteristics of simple operation, quick results, and good repeatability.
Characterization through X-ray Crystallography
X-ray crystallography stands as the pinnacle method for elucidating protein conformation. Despite its capability to furnish comprehensive structural insights into protein crystals, this technique necessitates a high-quality monocrystalline sample. The attainment of the requisite crystal structure poses challenges, particularly for large biomolecular proteins exhibiting intricate structures and flexibility. Furthermore, the crystallization process for a specific protein has the potential to induce alterations in its structure, resulting in non-identical states. Additionally, X-ray crystallography captures only a static snapshot of the protein's structure and is inadequate for discerning the solution conformation. Consequently, illustrating the relationship between macromolecular structure and function under physiological conditions becomes challenging. The intricate experimental procedures and protracted duration required also constitute limitations to the application of this technique.
Select Service
Nuclear Magnetic Resonance Spectroscopy Characterization
Nuclear Magnetic Resonance (NMR) is a routinely applied method in the analysis of chemical structure and reactivity characteristics, especially in determining protein and polypeptide structures in solutions. This methodology outstrips other physical analytical methods in its effectiveness. The principle of NMR allows for the examination of proteins with molecular weights less than 60kμ. Analysing the structure and properties of these proteins involves measuring their specific NMR spectral line parameters, transforming the primary data into distinct peaks using Fourier transformation, and subsequently assembling a spectrum composed of various peaks. This is followed by the utilisation of bioinformatics techniques to filter out spectra with specific structural features. These processes typically employ software such as NMRPipe and SPARKY, while the analysis of side-chain or backbone structures are performed using software like XEASY, DYANA, and GARANT.
NMR methods provide a valuable tool for studying protein kinetics on a broad temporal scale. In the instance of large molecular weight proteins and low signal sensitivity, orthogonal strategies such as solid-state NMR and site-specific amino acid labelling can be deployed to further probe protein structure, function, and dynamics. Such approaches enhance the depth and breadth of research capabilities within the field of structural biology.
Characterization via Cryo-Electron Microscopy
Cryo-electron microscopy (cryo-EM) embeds samples within a vitreous water environment through rapid, high-pressure freezing with liquid nitrogen, allowing for visualization of biomacromolecular structures in their native states. The ultrafast freezing can effectively capture cells at specific moments of physiological activity, therefore revealing structural features at these moments. Through studying instantaneous conformational changes across different functional states, this allows for investigation into the functions of biomolecules. Cryo-EM captures unstained biomolecular projections within their native states. By tilting the sample at various angles, an aggregate analysis of the collected data is performed. Depending on the distinct characteristics of the sample, different reconstruction techniques are employed to obtain the structures of the molecules. On this foundation, images of diverse components are observed, facilitating tracking of the assembly and dynamics of large biomolecules.
Advanced Protein Characterization Scheme
Apart from regular protein structure characterization approaches, Creative Proteomics also provides more comprehensive protein characterization analysis. This includes, O-glycosylation profiling, high resolution mass spectrometry using Electron Activated Dissociation (EAD) mode to identify isomers of amino acids, and also locating and confirming N-glycosylation of fucose.
References
- Nadler W M, Waidelich D, et al. MALDI versus ESI: the impact of the ion source on peptide identification. Journal of proteome research, 2017, 16(3): 1207-1215.
- Wen-Shan Z, Hai-Ming L, et al. Significance of Peptide Mapping in the Quality Control of Recombinant Human Parathyriod Hormone 1-34 Product. chinese pharmaceutical journal, 2006.
- RAO C M, ZHANG Y, HAN C M, et al. Peptide mapping analysis of Rewnbinant human interleuldn-11 by tryptic digestion. Acta Pharm Sin, 2000, 35(5): 378-380.