Comprehensive Post-Translational Modification (PTM) Analysis for Confident Biologic Development
Regulatory agencies require thorough characterization of product-related variants arising from post-translational modifications (PTMs), as these directly impact the safety, efficacy, and stability of protein therapeutics. Our PTM analysis services provide deep structural insights into glycosylation, phosphorylation, disulfide bond connectivity, deamidation, oxidation, and a broad range of modification types — delivered under an ICH-aligned quality framework to support your preclinical development programs.
Request a Custom PTM Analysis ProposalIn line with ICH Q6B Guidance for biologic characterization
Jump to Section
- Why PTM Analysis Matters
- PTM Service Categories
- Glycosylation Analysis
- Other Critical PTMs
- Instrument Platform
- Service Advantages
- Analysis Workflow
- FAQs
Why Comprehensive PTM Analysis Is Critical for Biologic Development
Post-translational modifications (PTMs) are covalent chemical alterations that occur during or after protein biosynthesis, profoundly influencing the structure, function, stability, and immunogenicity of therapeutic proteins. Unlike small-molecule drugs, biologics — monoclonal antibodies, fusion proteins, bispecific antibodies, antibody-drug conjugates, and recombinant enzymes — are manufactured in living cell systems. This inherent biological complexity gives rise to a diverse array of PTMs that must be thoroughly characterized to ensure product quality, batch-to-batch consistency, and regulatory compliance.
The ICH Q6B guideline explicitly requires the identification and characterization of product-related substances and impurities, including post-translational modifications, as part of the marketing application for biotechnological products. PTMs such as glycosylation, oxidation, deamidation, and disulfide bond scrambling can alter the pharmacokinetic profile, reduce target-binding affinity, or trigger unwanted immune responses in patients. Proactive, high-resolution PTM profiling during early-stage development enables informed candidate selection, process optimization, and risk mitigation before costly late-stage failures.
At Creative Proteomics, our protein characterization platform integrates state-of-the-art mass spectrometry, chromatographic separation, and orthogonal spectroscopic techniques to deliver a comprehensive view of the PTM landscape across your biologic pipeline. We bring deep expertise in characterizing complex modalities — including ADC, bispecific antibodies, PEGylated proteins, and fusion proteins — under an ICH-aligned analytical framework that bridges discovery-stage developability assessment to preclinical candidate selection.
Our PTM analysis portfolio addresses three fundamental characterization needs:
- PTM Identification & Site Mapping — precise localization of modification sites at the amino acid level using high-resolution MS/MS fragmentation
- PTM Quantification — relative and absolute quantitation of modification occupancy, including site-specific glycan profiling and oxidation/deamidation kinetics
- PTM Impact Assessment — correlating modification status with structural integrity, thermal stability, binding activity, and aggregation propensity
Comprehensive PTM Service Categories
Our analytical platform covers a broad spectrum of known and emerging PTM types relevant to biotherapeutic development. Each service category is supported by dedicated method development capabilities to address the specific analytical challenges posed by different modification chemistries.
Glycosylation
Key Modifications: N-glycans, O-glycans, sialic acid, glycopeptide profiling
Primary Technique: LC-MS/MS, MALDI-TOF, HILIC-FLD
Biological Relevance: Effector function, PK half-life, immunogenicity
Phosphorylation
Key Modifications: Ser/Thr/Tyr phosphorylation, phosphoproteomics
Primary Technique: TiO₂ enrichment + LC-MS/MS
Biological Relevance: Signaling pathway modulation, potency
Disulfide Bonds
Key Modifications: Disulfide bridge mapping, free sulfhydryl, scrambling
Primary Technique: Non-reduced peptide mapping LC-MS/MS
Biological Relevance: Structural integrity, stability, misfolding
Oxidation & Deamidation
Key Modifications: Met/Trp oxidation, Asn deamidation, Asp isomerization
Primary Technique: LC-MS/MS, peptide mapping
Biological Relevance: Stability, aggregation, loss of activity
Acetylation
Key Modifications: N-terminal acetylation, Lys acetylation
Primary Technique: LC-MS/MS, immunoaffinity enrichment
Biological Relevance: Stability, charge variants, immunogenicity
Ubiquitination & SUMOylation
Key Modifications: Ubiquitin remnant (diGly), SUMO1/2/3 chains
Primary Technique: K-ε-GG enrichment + LC-MS/MS
Biological Relevance: Protein degradation, cellular signaling
Methylation & Alkylation
Key Modifications: Lys/Arg methylation, ethylation, carboxymethylation
Primary Technique: LC-MS/MS, ETD/ECD fragmentation
Biological Relevance: Epigenetic regulation, charge heterogeneity
Lipidation
Key Modifications: Palmitoylation, myristoylation, prenylation
Primary Technique: Acyl-biotin exchange + LC-MS/MS
Biological Relevance: Membrane association, protein trafficking
S-Nitrosylation
Key Modifications: Cys S-nitrosylation, S-glutathionylation
Primary Technique: Biotin-switch assay + LC-MS/MS
Biological Relevance: Redox regulation, signaling
PEGylation Characterization
Key Modifications: PEG attachment sites, PEG heterogeneity
Primary Technique: LC-MS/MS, SEC-MALS
Biological Relevance: PK extension, immunogenicity risk
In-Depth Glycosylation Analysis for Biotherapeutics
Glycosylation is widely recognized as the most structurally complex and functionally significant PTM affecting therapeutic proteins. The specific glycan structures attached at each glycosylation site — their composition, branching pattern, linkage configuration, and sialylation status — collectively determine the circulating half-life, Fc receptor binding affinity, antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and immunogenicity profile of monoclonal antibody therapeutics.
Our glycosylation analysis service suite encompasses multiple analytical levels to address the full complexity of glycoprotein characterization:
Intact Glycoform Profiling
Intact mass analysis under native or denaturing conditions provides a global snapshot of the glycoform distribution, enabling rapid assessment of batch-to-batch glycosylation consistency and identification of unexpected glycan variants. We deploy both ESI-TOF and MALDI-TOF platforms for complementary coverage of low- and high-mass glycoforms.
Released Glycan Analysis
Enzymatic release (PNGase F for N-glycans, β-elimination or enzymatic release for O-glycans) followed by MALDI-TOF MS profiling, HILIC-FLD with 2-AB labeling, or LC-MS/MS provides detailed structural characterization of individual glycan species, including monosaccharide composition, branching topology, and sialic acid linkage (α2,3 vs α2,6).
Site-Specific Glycopeptide Analysis
The gold-standard approach for therapeutic glycoproteins: LC-MS/MS analysis of intact glycopeptides from a proteolytic digest simultaneously reveals the glycan structure and its precise attachment site within the protein sequence. Our platform supports both data-dependent acquisition (DDA) and data-independent acquisition (DIA) workflows for comprehensive site-specific glycosylation profiling.
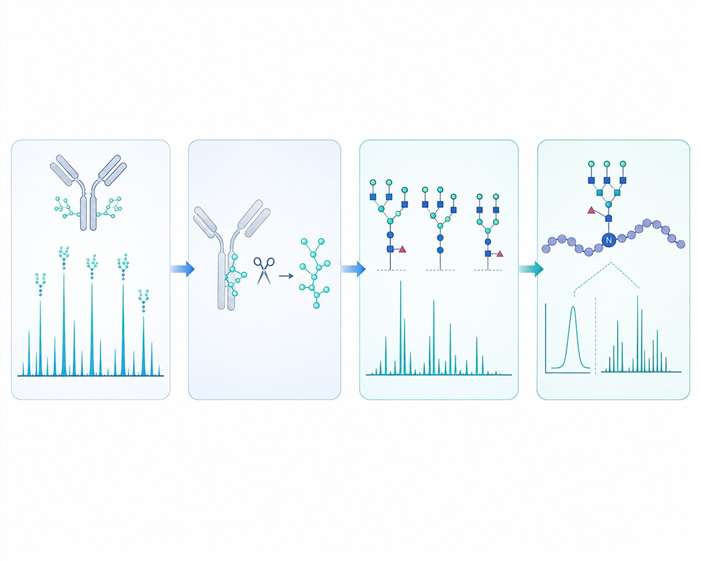
Comprehensive Coverage of Critical PTMs in Biologic Development
Beyond glycosylation, our analytical platform addresses the full breadth of product-related PTM variants that regulatory agencies expect to be characterized during biologic development. Each PTM type requires specialized sample preparation, enrichment strategies, and mass spectrometric acquisition methods to achieve reliable detection and accurate quantitation.
| PTM Type | Analytical Strategy | Key Information Delivered |
|---|---|---|
| Phosphorylation | Immobilized metal affinity chromatography (IMAC) or TiO₂ enrichment → LC-MS/MS with HCD/ETD fragmentation | Site-specific phosphorylation stoichiometry, kinase-substrate inference, signaling pathway impact |
| Disulfide Bridges & Free Thiols | Non-reduced peptide mapping with alkylation of free cysteines → LC-MS/MS; differential alkylation for disulfide/scrambling analysis | Native disulfide connectivity map, free sulfhydryl content, scrambling hotspots under stress |
| Deamidation & Isomerization | Trypsin/Lys-C peptide mapping → LC-MS/MS with targeted extracted ion chromatogram (XIC) quantitation | Site-specific deamidation rate (Asn→Asp/isoAsp), Asp isomerization kinetics under accelerated stability |
| Oxidation | Peptide mapping → LC-MS/MS (Met oxidation: +16 Da; Trp oxidation: +16, +32, +48 Da) | Oxidation hotspot identification, forced degradation kinetics, formulation impact assessment |
| Acetylation | Trypsin digestion → LC-MS/MS with or without acetyl-Lys enrichment | N-terminal acetylation confirmation, Lys acetylation site mapping, charge variant correlation |
| Methylation & Alkylation | LC-MS/MS with ETD for labile modification retention; HCD for quantitative methyl occupancy | Methylation site identification (mono/di/tri-methyl), ethylation/carboxymethylation from process-related impurities |
| Ubiquitination & SUMOylation | Trypsin digestion (diGly remnant enrichment for ubiquitin) → LC-MS/MS; SUMO remnant antibody enrichment | Ubiquitination site mapping, ubiquitin chain topology (K48 vs K63), SUMO paralog identification |
| PEGylation | Intact mass analysis → LC-MS; PEGylated peptide mapping → LC-MS/MS; SEC-MALS for size distribution | PEG attachment site verification, PEG chain length heterogeneity, free PEG quantitation |
| Lipidation | Acyl-biotin exchange (ABE) for palmitoylation; metabolic labeling for myristoylation → LC-MS/MS | S-acylation (palmitoylation) site identification, N-myristoylation confirmation, prenylation profiling |
| S-Nitrosylation & S-Glutathionylation | Biotin-switch assay (BST) or direct MS-based approaches → LC-MS/MS | Nitrosylated cysteine identification, S-glutathionylation sites, redox-sensitive structural elements |
| Glycation | Boronate affinity enrichment → LC-MS/MS; neutral loss scanning for early glycation products | Glycation site mapping (Lys/Arg), early-stage (Amadori) vs advanced glycation end-product (AGE) differentiation |
| Hydroxylation | LC-MS/MS with high-resolution accurate mass for +16 Da shift localization | Proline/lysine hydroxylation mapping, collagen-domain modification assessment |
| Proteolytic Cleavage | Intact mass analysis + LC-MS/MS peptide mapping with N/C-terminal peptide identification | Clipped variant identification, fragmentation hotspot mapping, cathepsin/caspase cleavage characterization |
For each PTM category, our analytical scientists develop and qualify fit-for-purpose methods that balance depth of characterization with project timelines. We routinely combine multiple orthogonal techniques — including HCD, ETD, and EThcD fragmentation — to achieve unambiguous modification site assignment. Dedicated service pages for each PTM type provide deeper technical details on method parameters, analytical strategies, and application-specific workflows tailored to your biologic modality.
Instrument Platform for PTM Characterization
Reliable PTM analysis demands instrumentation capable of high-resolution precursor mass measurement, sensitive MS/MS fragmentation, and robust chromatographic separation. Creative Proteomics operates a comprehensive suite of mass spectrometers and separation systems to address the analytical diversity of biologic PTM characterization:
- Orbitrap-based platforms (Thermo Fisher Q Exactive HF-X, Orbitrap Eclipse) — high-resolution accurate mass (HRAM) for intact protein analysis and top-down PTM characterization
- Q-TOF systems (Agilent 6520, Sciex TripleTOF 6600) — fast scanning for comprehensive LC-MS/MS glycopeptide and phosphopeptide analysis
- MALDI-TOF/TOF (Bruker UltrafleXtreme) — rapid glycan profiling, intact mass screening, and in-source decay for labile modification analysis
- Triple Quadrupole MS (Sciex 6500+) — targeted MRM-based PTM quantitation for site-specific modification kinetics and multi-attribute monitoring (MAM)
- NanoLC and UHPLC systems (Thermo EASY-nLC, Waters ACQUITY UPLC) — high-resolution peptide separation with HILIC, RP, and mixed-mode chromatography for glycopeptide and modified peptide analysis



Service Advantages
Deep PTM Expertise
Our scientists have extensive experience characterizing 15+ PTM classes across diverse biologic modalities — from monoclonal antibodies to fusion proteins, ADCs, bispecifics, and gene therapy products. We bring domain-specific knowledge to each project, ensuring appropriate method selection and data interpretation.
Orthogonal Analytical Approaches
We routinely deploy complementary techniques — HCD, ETD, EThcD, UV-Vis, fluorescence, SEC-MALS — to resolve ambiguous PTM assignments and provide multi-dimensional evidence for modification identification and structural impact assessment.
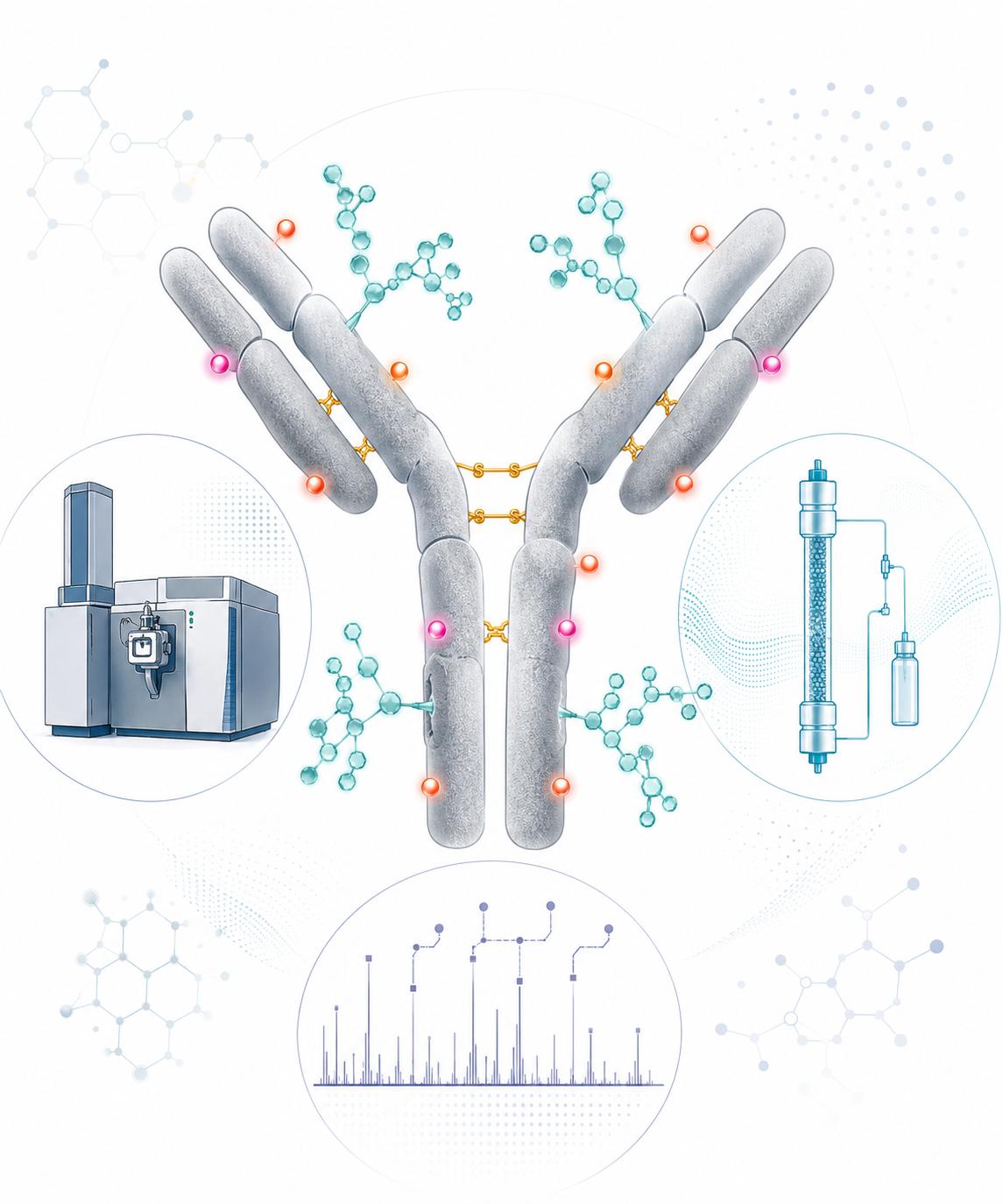
ICH-Aligned Quality Framework
All PTM characterization projects are executed under a quality system aligned with ICH Q2(R1) method validation principles and ICH Q6B product characterization expectations, supporting seamless data transition from discovery-stage profiling to regulatory submission.
Integrated Multi-Attribute Monitoring
Our advanced LC-MS/MS workflows support multi-attribute method (MAM) implementation — enabling simultaneous monitoring of multiple PTM attributes in a single analytical run for efficient batch comparability and stability assessment.
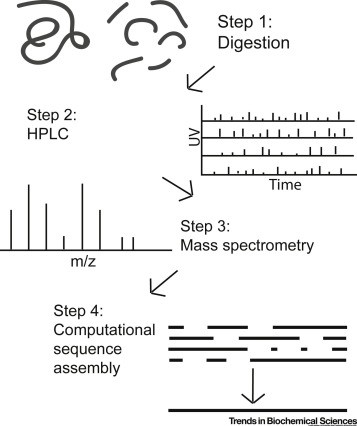
PTM Analysis Workflow
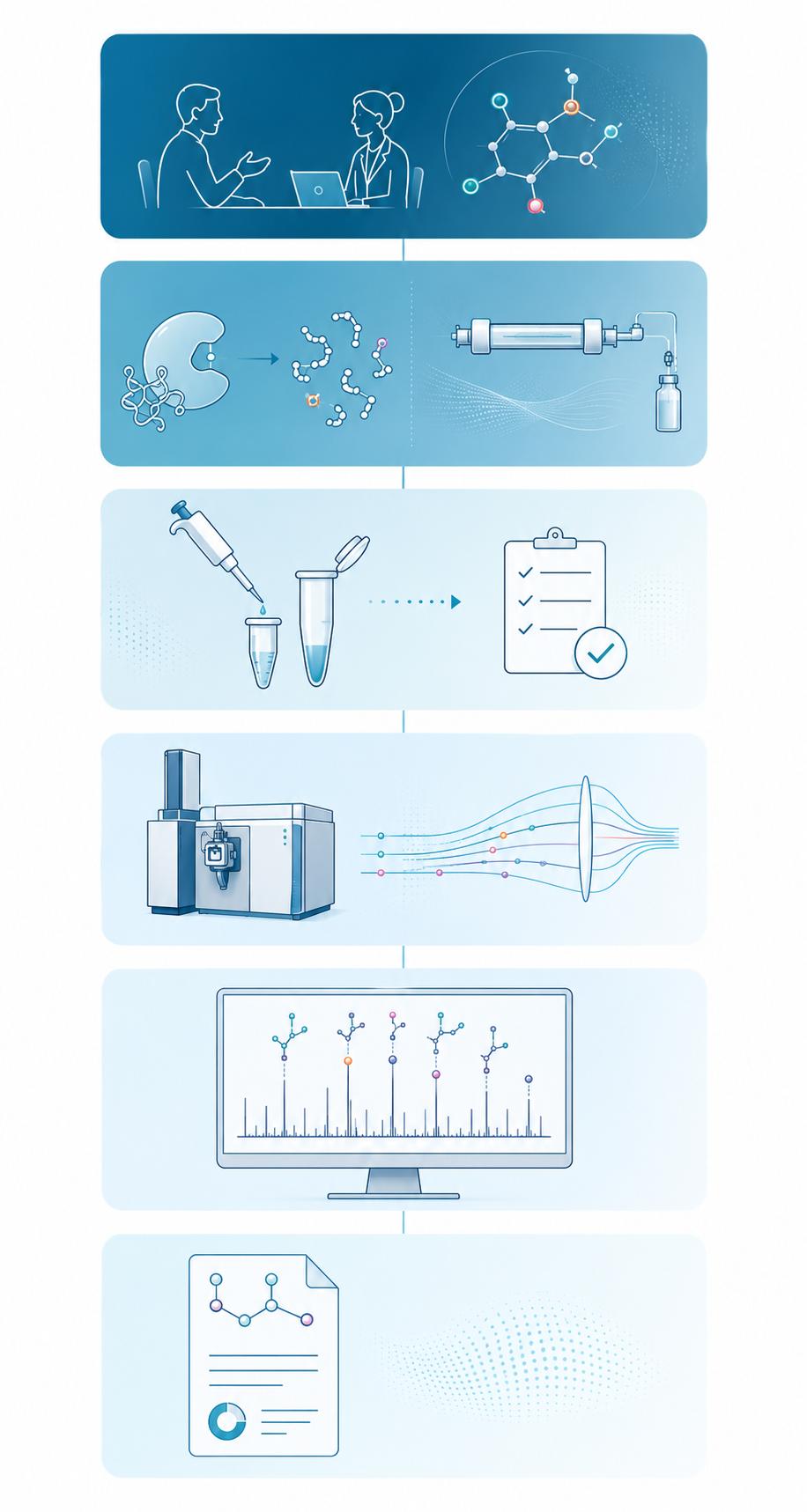
- 1
Project Consultation & PTM Scope Definition
We begin by understanding your biologic modality, development stage, and regulatory expectations. Together we define which PTM attributes require characterization, the depth of analysis needed, and project timelines aligned with your development milestones.
- 2
Custom Method Development & Qualification
Based on the PTM scope, we design fit-for-purpose analytical methods — selecting appropriate digestion protocols (trypsin, Lys-C, chymotrypsin, or multi-enzyme), enrichment strategies (IMAC, TiO₂, lectin affinity, boronate affinity), and LC-MS/MS acquisition parameters.
- 3
Sample Preparation & Quality Control
We provide detailed sample submission guidelines for your specific PTM analysis. Upon sample receipt, we perform integrity checks, quantify total protein, and execute the validated sample preparation workflow with appropriate process controls and blanks.
- 4
LC-MS/MS Data Acquisition
High-resolution MS data are acquired on optimized instrument platforms using targeted and discovery-based acquisition methods. Each acquisition includes system suitability standards, retention time calibration, and mass accuracy validation.
- 5
PTM Identification & Quantitation
MS/MS data are analyzed using established search engines (Proteome Discoverer, Mascot, Byonic) with appropriate PTM variable modifications and FDR-controlled validation. Site-specific quantitation is performed via XIC peak integration or isotopic labeling as applicable.
- 6
Comprehensive Reporting & Expert Interpretation
We deliver a detailed technical report including modification site maps, quantitative data, annotated MS/MS spectra, and biological context interpretation. Our scientists are available for follow-up discussions and regulatory query support.
Case Study: LC-MS/MS-Based Identification of a Novel PTM in Monoclonal Antibody Stability Assessment
In a recent investigation focused on monoclonal antibody formulation stability, researchers applied high-resolution LC-MS/MS peptide mapping to characterize an unexpected modification emerging during accelerated stability studies. The study, conducted on a therapeutic IgG1 monoclonal antibody (mAb X), identified sorbitol — a common excipient used in biotherapeutic formulations — covalently conjugated to glutamic acid residues via ester bond formation under acidic storage conditions.
Key findings:
- LC-MS/MS peptide mapping with high-resolution Orbitrap MS revealed a +182 Da mass shift corresponding to sorbitol attachment on specific glutamic acid residues
- Sites of modification were localized to solvent-exposed Glu residues within the complementarity-determining regions (CDRs) of the antibody Fab domain
- The sorbitol esterification was found to be pH-dependent, with maximum occurrence observed at pH 4.5–5.0, a typical range for liquid mAb formulations
- Kinetic analysis demonstrated time-dependent accumulation of the modification over a 6-month accelerated stability period at 25°C and 40°C
- Molecular dynamics simulations suggested that the modification could induce localized conformational perturbations in the CDR loops, potentially affecting antigen-binding affinity
This case highlights the importance of comprehensive PTM profiling extending beyond the conventional modification panel (oxidation, deamidation, glycation), and demonstrates how high-resolution LC-MS/MS can detect unexpected product-related variants that may impact product quality and stability.
Source: [1]
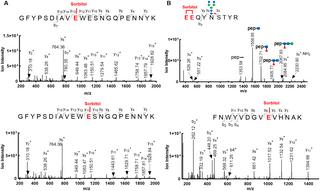
Figure from Yu et al. (2024) showing LC-MS/MS identification of sorbitol esterification on glutamic acid residues (CC BY 4.0)
Frequently Asked Questions About PTM Analysis
What sample amount is required for PTM analysis by LC-MS/MS?
For standard peptide mapping-based PTM analysis, 50–200 μg of purified protein is typically sufficient for comprehensive modification profiling. For phosphopeptide enrichment or ubiquitination analysis, larger amounts (200–500 μg) may be required to compensate for the low stoichiometry of these modifications. We recommend contacting our scientists to discuss the specific PTM panel and sample availability for your project.
Can you detect and quantify multiple PTMs simultaneously in a single analysis?
Yes. Using multi-attribute method (MAM) workflows on high-resolution Orbitrap platforms, we can simultaneously monitor glycosylation, oxidation, deamidation, isomerization, and other PTM attributes in a single LC-MS/MS peptide mapping experiment. This approach is particularly valuable for comparability studies, stability monitoring, and batch release testing where multiple quality attributes must be tracked concurrently.
How do you distinguish between different PTM types that produce the same mass shift?
We address this challenge through a combination of strategies: (1) high-resolution accurate mass measurement to distinguish isobaric modifications when possible; (2) orthogonal fragmentation techniques (ETD vs HCD) that provide complementary structural information; (3) targeted MS/MS acquisition with diagnostic fragment ions; and (4) independent confirmation using alternative enzymatic digestion or chemical derivatization approaches when ambiguity persists.
What is the turnaround time for a typical PTM characterization project?
Project timelines depend on the scope and complexity of the PTM panel required. For a standard peptide mapping-based modification assessment (oxidation, deamidation, glycosylation profiling), results are typically available within 2–3 weeks from sample receipt. For projects requiring specialized enrichment (phosphorylation, ubiquitination) or method development, timelines may extend to 4–6 weeks. We provide a detailed project timeline during the initial consultation phase.
Can you perform PTM analysis on formulated drug product or stressed samples?
Absolutely. We routinely analyze PTM profiles across a variety of sample types including formulated drug substance, drug product, stressed/forced degradation samples, and stability study time points. We have established protocols for buffer exchange and sample cleanup to remove formulation excipients that may interfere with LC-MS/MS analysis, ensuring robust PTM detection even in complex formulation matrices.
References
- Yu B, Williams S, Yang Z, Young G. Identification of sorbitol esterification of glutamic acid by LC-MS/MS in a monoclonal antibody stability assessment. PLOS ONE. 2024;19(5):e0295735.
- Senini I, Tengattini S, Rinaldi F, et al. Direct glycosylation analysis of intact monoclonal antibodies combining ESI MS of glycoforms and MALDI-in source decay MS of glycan fragments. Commun Chem. 2024;7(1):203.
- Mojumdar A, et al. Advances in mass spectrometry-based approaches for characterizing monoclonal antibodies: resolving structural complexity and analytical challenges. J Anal Sci Technol. 2024;15:23.





Ready to Characterize Your Biologic's PTM Profile?
Request Your Custom PTM Analysis PlanPartner with our expert team to design a comprehensive PTM characterization strategy that delivers the analytical depth and regulatory confidence needed to advance your biologic development program.
For Research Use Only. Not for diagnostic procedures.
