First introduced by Kaj Ulrik Linderstrom-Lang at Stanford in 1952, protein secondary structure is the three dimensional form of local segments of proteins and refers to the pattern of hydrogen bonds between the carboxyloxygen atoms and amino hydrogen in the peptide backbone. Protein secondary structure elements is an intermediate before the protein folds into its three dimensional tertiary structure and is typically spontaneously formed.
Though beta turns and omega loops occur as well, the α-helix and the ß-sheet are the most common types of secondary structure. α-helix is a right hand-spiral conformation in which every backbone N−H group donates a hydrogen bond to the backbone C=O group of the amino acid that is four down the chain. The side-chain substituents of the amino acids fit in beside the N-H groups. The polypeptide chain is pulled by hydrogen bonds into a helical structure resembling a curled ribbon, with each turn of the helix containing 3.6 amino acids. The α-helix the most regular, most prevalent and the most predictable from sequence. The β sheets is the second most abundant secondary structure, and arises when several β-strands, stretched segments of the polypeptide chain are kept together by a network of hydrogen bonds. Rather than within strands (intra-strand), the hydrogen bonding in a ß-sheet is between strands (inter-strand). Depending on whether the strand directions (N-terminus to C-terminus) are the same or opposite, the two strands can be either parallel or anti-parallel. Like the α helix, owing to its hydrogen bonds and the Van der Waals forces that exist because of the close proximity of the residues in the structure, the β pleated sheet is stable. The anti-parallel ß-sheet is more stable due to the more well-aligned hydrogen bonds. Though often being hypothesized as important protein folding intermediates, other extended structures such as the polyproline helix and alpha sheet are rare in native state proteins while more "regular" secondary structure elements such as α-helix and the ß-sheet are linked by tight turns and loose, flexible loops link.
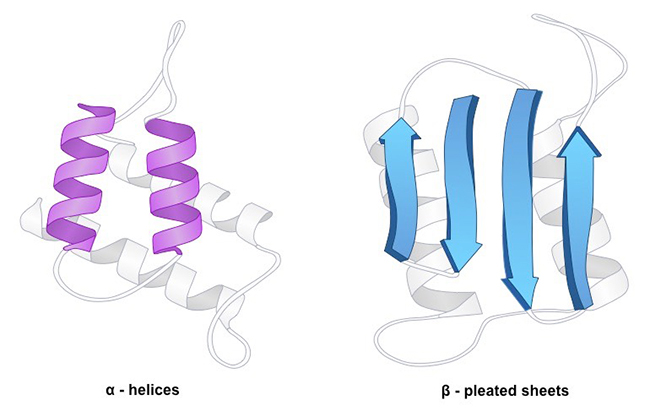 Fig1. Protein Secondary Structure
Fig1. Protein Secondary StructureAmino acids vary in their ability to form the various secondary structure elements. Known as "helix breakers", proline and glycine disrupt the regularity of the α helical backbone conformation, though they are commonly found in turns. Proteins include methionine, alanine, leucine, glutamate and lysine prefer to adopt helical conformations, while β-branched amino acids (isoleucine, valine, and threonine) and the large aromatic residues (tryptophan, tyrosine and phenylalanine) prefer to adopt β-strand conformations in contrast. However, these preferences are not strong enough to predict secondary structure reliably from sequence alone.
The secondary structure is an important level in the hierarchical classification of protein structure and it is used to identify protein features for fold recognition. Secondary structure is very important to protein in the evolution, size and geometry selection of the secondary structure motifs. Circular dichroism (CD) is an excellent method for rapidly evaluation of the secondary structure, folding and binding properties of proteins. Secondary Structure Analysis services at Creative Proteomics are as below.
- Circular Dichroism Spectra (Far UV)
- Fluorescence Spectroscopy
- Nuclear Magnetic Resonance (NMR) Spectroscopy
- FT-infrared Spectroscopy
- Native Mass Spectrometry Analysis
For more details, please feel free to contact us or send inquiry directly.





