Phosphorylation Protein Analysis for Confident Biotherapeutic Development
Phosphorylation of recombinant therapeutic proteins — arising during cell culture production, purification, or formulation — introduces product-related variants that can alter target-binding affinity, modulate Fc effector function, and generate charge heterogeneity. These modifications are routinely overlooked in standard characterization panels yet have measurable impact on product quality, batch consistency, and regulatory risk profiles. Our phosphorylation analysis service delivers unambiguous phosphosite identification, precise site localization, and stoichiometric quantitation using modality-specific LC-MS/MS strategies with orthogonal fragmentation — executed under an ICH-aligned quality framework to support your preclinical and CMC development programs.
Request a Custom Phosphorylation Analysis PlanIn line with ICH Q6B Guidance for biologic characterization
Jump to Section
- Why Phosphorylation Analysis Matters
- Service Capabilities
- Analysis by Modality
- Analytical Approaches
- Service Advantages
- Instrument Platform
- Workflow
- Case Study
- FAQs
Why Phosphorylation Analysis Matters in Biologic Development
Phosphorylation — the reversible addition of a phosphate group (−PO₃²⁻, +79.966 Da) to serine, threonine, or tyrosine residues — is the most abundant signaling-associated post-translational modification in mammalian cells. In recombinant therapeutic proteins produced in CHO or HEK293 expression systems, phosphorylation occurs during biosynthesis as a direct consequence of cell signaling states, culture conditions, and metabolic stress. Unlike oxidation or deamidation, phosphorylation sites in therapeutic proteins are routinely undetected in standard peptide mapping workflows because phosphopeptides ionize poorly in positive-ion mode and the labile phosphate group dissociates under conventional CID fragmentation, making unambiguous identification inherently challenging without dedicated methods.
For CMC and quality teams, the practical concern is threefold:
- Regulatory risk: ICH Q6B requires characterization of product-related substances. Undetected phosphorylation variants can surface during later comparability or stability studies, triggering unexpected regulatory queries and potentially delaying development timelines
- Charge heterogeneity impact: Each phosphate group introduces a −2 charge shift at physiological pH, directly contributing to acidic variant profiles observed in ion exchange chromatography. Without MS-based confirmation, the root cause of these variants remains speculative
- Process sensitivity: Phosphorylation levels fluctuate with cell culture parameters — dissolved oxygen, nutrient limitation, harvest timing — making phosphorylation patterns sensitive indicators of process consistency and potential critical quality attributes (CQAs) requiring monitoring in a multi-attribute method (MAM) framework
Our approach addresses these challenges through modality-specific method development: the digestion strategy, enrichment chemistry, and MS fragmentation parameters are optimized for each biologic class, not applied as a one-size-fits-all workflow. This ensures that phosphorylation variants are detected, localized, and quantified with the analytical confidence required for CMC decision-making and regulatory submission support. Our comprehensive PTM analysis platform integrates these capabilities within a broader ICH-aligned characterization framework spanning glycosylation, oxidation, deamidation, and disulfide bond analysis.
Phosphorylation Analysis Service Capabilities
Our analytical platform delivers end-to-end phosphorylation characterization — from discovery-level phosphosite mapping through targeted absolute quantitation — across all major biotherapeutic modalities.
Phosphosite Discovery Mapping
Unbiased identification of all phosphorylation sites across your protein sequence using TiO₂ or IMAC enrichment with HCD/ETD decision-tree fragmentation. Delivers a comprehensive phosphosite inventory with localization scores.
Site-Specific Stoichiometric Quantitation
Absolute quantitation of phosphorylation occupancy at individual sites using targeted PRM with stable isotope-labeled synthetic phosphopeptide standards (AQUA). Determines precise site occupancy percentages and temporal kinetics.
Multi-Attribute Method (MAM) Monitoring
Simultaneous tracking of phosphorylation across multiple sites alongside oxidation, deamidation, and glycosylation in a single LC-MS/MS run. Ideal for batch comparability, stability monitoring, and process consistency assessment.
Top-Down Phosphorylation Profiling
Intact protein-level analysis using ETD and UVPD fragmentation on Orbitrap Eclipse. Reveals phosphorylation isoform distribution and combinatorial phosphosite patterns without digestion, preserving the native modification context.
2D Phosphopeptide Mapping
Deep coverage of multiply phosphorylated peptides using SCX or HILIC fractionation prior to TiO₂ enrichment and EThcD analysis. Enables identification of phosphorylation crosstalk and cooperative site occupancy patterns.
Phosphorylation Variant Confirmation
Orthogonal validation of ambiguous phosphosite assignments using alkaline phosphatase treatment (biochemical confirmation by mass shift reversal), synthetic phosphopeptide co-elution, and multi-enzyme digestion strategies.
Phosphorylation Analysis Across Biologic Modalities
Phosphorylation analysis is not a one-method-fits-all service. Each biologic modality presents distinct structural contexts where phosphorylation can occur, requiring tailored analytical strategies for reliable detection and unambiguous site localization. Our scientists design modality-specific workflows that account for differences in protein architecture, expression host, and the phosphorylation motifs most relevant to each drug class.
Monoclonal Antibodies (IgG₁, IgG₄)
Most Common ModalityPhosphorylation occurs on surface-exposed Ser/Thr in CDR loops and Fc regions. CDR phosphorylation directly risks antigen-binding affinity; Fc phosphorylation modulates FcγR binding and ADCC/CDC activity. Requires multi-enzyme digestion (trypsin + Lys-C + chymotrypsin) for full sequence coverage of both Fab and Fc domains.
Key analytical consideration: CDR phosphopeptides often hydrophobic — HILIC fractionation recommended for comprehensive coverage.
Trypsin/Lys-C digestion HCD + EThcD TiO₂ enrichmentBispecific Antibodies
Higher ComplexityMultiple chain assembly (2–4 distinct polypeptide chains) creates more potential phosphorylation sites. Linker regions between domains are particularly susceptible, as the case study below demonstrates. Requires chain-specific peptide mapping to assign phosphorylation to the correct chain.
Key analytical consideration: Identical sequence motifs in different chains require thermolysin or multi-enzyme digestion for unambiguous chain assignment.
Linker-focused analysis Multi-enzyme mapping Intact MS confirmationFc-Fusion & Albumin-Fusion Proteins
Linker RiskGlycine-serine (G₄S)ₙ linkers widely used in fusion constructs create CK2 kinase consensus motifs (SSSGSSSG), making them inadvertent phosphorylation hotspots. Phosphorylation in linkers does not directly affect target binding but can introduce charge heterogeneity and potentially alter pharmacokinetics.
Key analytical consideration: Linker peptides are small and hydrophilic — standard C18 columns may not retain them well; HILIC or mixed-mode chromatography recommended.
Linker phosphorylation HILIC chromatography ETD for localizationAntibody-Drug Conjugates (ADCs)
Conjugation ComplexityPhosphorylation analysis of ADCs requires deconvoluting the phosphorylation signal from the conjugated payload signal. The drug-antibody ratio (DAR) distribution must be accounted for during intact mass analysis. Payload-containing peptides may co-elute with phosphopeptides, requiring additional fractionation.
Key analytical consideration: ADC payloads are hydrophobic — deglycosylation and reduction are strongly recommended before LC-MS/MS to reduce sample complexity.
Deglycosylation DAR normalization High-resolution MSRecombinant Enzymes & Cytokines
Activity-CriticalPhosphorylation at or near the active site of recombinant enzymes can directly reduce or abolish catalytic activity — a critical quality attribute. For cytokines and growth factors, phosphorylation near receptor-binding interfaces can alter potency. Requires particularly high sequence coverage (≥95%) to ensure active-site regions are assessed.
Key analytical consideration: Active-site peptides are often large and hydrophobic — multi-enzyme digestion essential to achieve the sequence coverage required for regulatory confidence.
≥95% sequence coverage Activity correlation Multi-enzyme digestionPEGylated Proteins
Unique InterferencePEG polymers suppress ionization of conjugated peptides in MS, making phosphorylation detection on PEGylated chains extremely challenging. Requires enzymatic or chemical de-PEGylation prior to LC-MS/MS analysis, with careful controls to distinguish phosphorylation from PEG fragmentation artifacts.
Key analytical consideration: De-PEGylation efficiency must be verified; residual PEG interferes with reversed-phase chromatography and suppresses phosphopeptide ionization.
De-PEGylation step Ionization suppression check Control standardsAnalytical Approaches for Phosphorylation Characterization
Reliable phosphorylation analysis presents unique analytical challenges. Phosphopeptides ionize with lower efficiency than their unmodified counterparts, phosphorylation is often substoichiometric (0.5–15% occupancy), and the phosphate group is labile under conventional collision-induced dissociation (CID), making site localization difficult. Our platform addresses these challenges through a suite of complementary strategies.
| Analysis Type | Enrichment Strategy | MS Fragmentation | Key Information Delivered |
|---|---|---|---|
| Phosphosite Discovery Mapping | TiO₂ or IMAC (Fe³⁺/Ga³⁺) enrichment | HCD + ETD decision-tree fragmentation | Comprehensive phosphosite inventory, sequence coverage of phosphorylation across the entire protein |
| Site-Specific Stoichiometric Quantitation | TiO₂ enrichment with spiked synthetic phosphopeptide standards (AQUA/PRM) | Targeted PRM (parallel reaction monitoring) on Orbitrap platforms | Absolute stoichiometry per phosphosite, site occupancy percentage, temporal phosphorylation kinetics |
| Multi-Attribute Method (MAM) | No enrichment (direct peptide mapping with XIC extraction) | HCD on high-resolution Orbitrap (Q Exactive HF-X, Orbitrap Eclipse) | Simultaneous monitoring of phosphorylation alongside oxidation, deamidation, and glycosylation in a single LC-MS/MS run |
| Top-Down Phosphoproteomics | No digestion; intact protein level analysis | ETD on Orbitrap Eclipse with UVPD optional | Phosphorylation isoform distribution at the intact protein level, combinatorial phosphosite patterns |
| 2D Phosphopeptide Mapping | SCX or HILIC fractionation plus TiO₂ enrichment | EThcD for unambiguous site localization | Deep coverage of multiply phosphorylated peptides, phosphorylation crosstalk identification |
Our analytical scientists select the most appropriate enrichment and fragmentation strategy based on your biologic modality, phosphorylation level of interest, and quantitative accuracy requirements. For projects requiring orthogonal confirmation, we routinely integrate complementary MS-based approaches to resolve ambiguous site assignments.
Service Advantages
Orthogonal Fragmentation for Unambiguous Site Localization
Phosphorylation site assignment by conventional CID alone is unreliable — the labile phosphate group dissociates before peptide backbone fragmentation occurs. We deploy decision-tree-driven HCD + ETD/EThcD acquisition on Orbitrap platforms, ensuring that every identified phosphosite is confirmed by complementary fragmentation evidence. This approach eliminates ambiguous assignments and provides the analytical confidence required for regulatory submission.
Multi-Platform Enrichment Portfolio
No single enrichment method captures the full phosphoproteome. Our platform supports TiO₂ chromatography, IMAC (Fe³⁺/Ga³⁺), and sequential enrichment strategies, enabling tailored coverage for your specific phosphorylation level of interest — from low-abundance tyrosine phosphorylation to multiply phosphorylated peptide species.
ICH-Aligned Data Package for CMC Confidence
Every phosphorylation characterization project is executed under a quality system aligned with ICH Q2(R1) method validation principles. Our deliverable package includes annotated MS/MS spectra with localization scores, extraction ion chromatograms for stoichiometric quantitation, and a comprehensive method summary — providing the analytical evidence chain needed for CMC documentation and regulatory query support.
Stoichiometric Quantitation with Absolute Accuracy
Beyond qualitative site mapping, we deliver site-specific phosphorylation stoichiometry using either label-free XIC quantitation (phosphorylated vs non-phosphorylated peptide pair comparison) or targeted PRM with stable isotope-labeled synthetic phosphopeptide standards — enabling precise determination of site occupancy percentages and temporal phosphorylation kinetics.
Instrument Platform for Phosphorylation Analysis
Reliable phosphorylation analysis demands instrumentation capable of high-resolution accurate mass measurement, sensitive MS/MS acquisition with orthogonal fragmentation capabilities, and robust nanoLC separation for phosphopeptide analysis. Creative Proteomics operates a dedicated suite of mass spectrometry platforms configured and optimized for phosphorylation characterization workflows:
- Orbitrap Eclipse Tribrid — ETD, HCD, and UVPD fragmentation options for unambiguous phosphosite localization; decision-tree acquisition for automated fragmentation mode selection based on precursor charge state and m/z
- Q Exactive HF-X — high-speed HCD for deep phosphoproteome coverage in data-dependent acquisition (DDA) and parallel reaction monitoring (PRM) modes for targeted phosphosite quantitation
- ZenoTOF 7600 — fast scanning with EAD (electron-activated dissociation) for complementary fragmentation to confirm phosphorylation site assignments on multiply phosphorylated peptides
- EASY-nLC 1200 — nanoflow UHPLC with integrated analytical and trap columns for high-sensitivity phosphopeptide separation; compatible with HILIC and reversed-phase chromatography modes



Phosphorylation Analysis Workflow
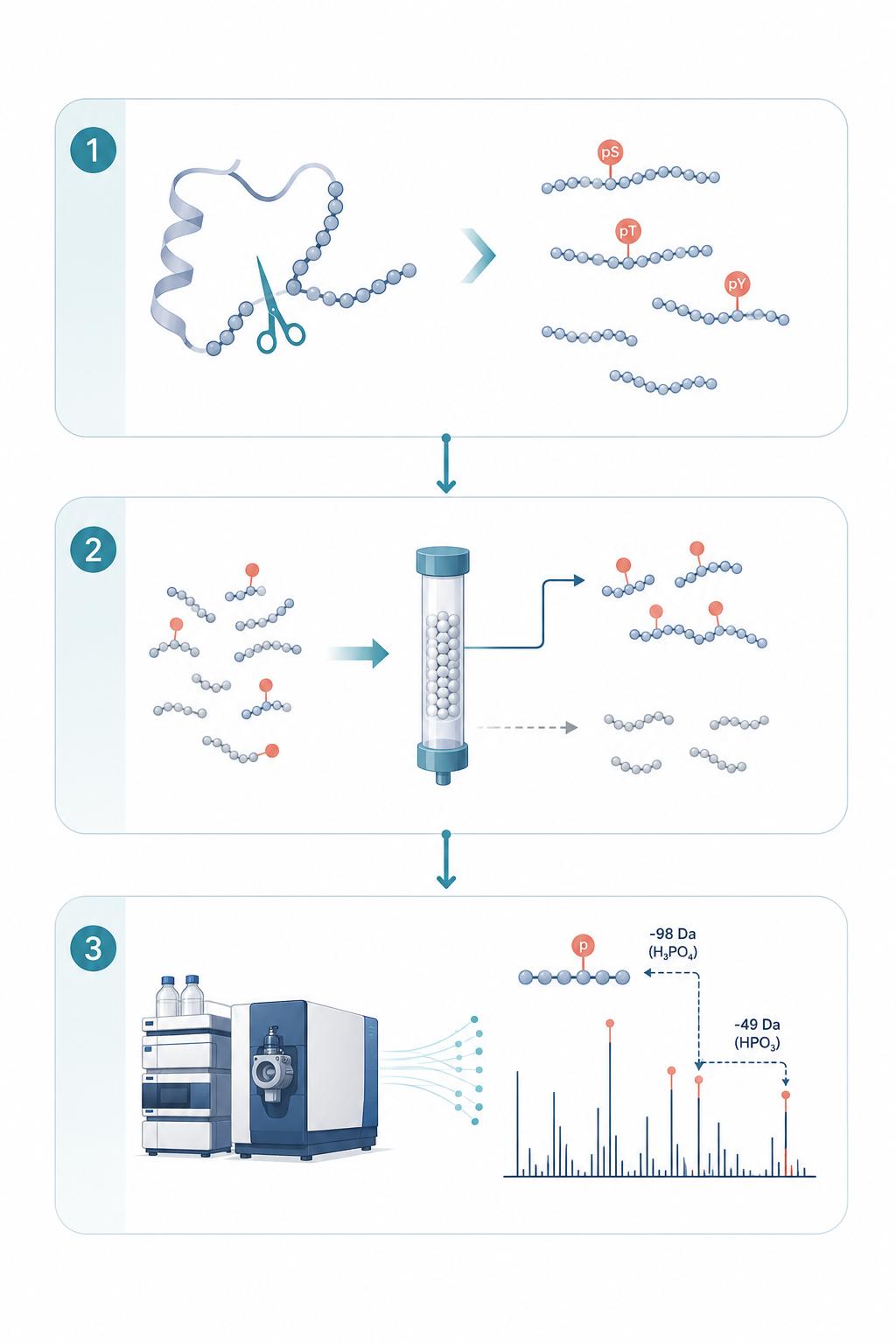
- 1
Project Consultation & Analytical Design
We discuss your biologic modality, known or suspected phosphorylation sites, expression host (CHO vs HEK vs other), and the regulatory context of your development program to define the optimal enrichment strategy, digestion protocol, and MS acquisition parameters.
- 2
Sample Preparation & Enzymatic Digestion
Your protein sample undergoes controlled proteolytic digestion (trypsin, Lys-C, chymotrypsin, or multi-enzyme combination based on modality) with phosphatase inhibitor cocktails to preserve native phosphorylation status. Process blanks and control standards are included for quality assurance.
- 3
Phosphopeptide Enrichment
Phosphopeptides are selectively enriched using TiO₂ chromatography, IMAC (Fe³⁺ or Ga³⁺), or sequential enrichment strategies optimized for your phosphorylation level of interest — from low-abundance tyrosine to multiply-phosphorylated species.
- 4
LC-MS/MS Data Acquisition
Enriched phosphopeptide fractions are analyzed by nanoLC-MS/MS on high-resolution Orbitrap or ZenoTOF platforms. We deploy orthogonal fragmentation (HCD for identification, ETD/EThcD/EAD for unambiguous site localization) in a decision-tree acquisition framework.
- 5
Data Analysis & Phosphosite Validation
MS/MS data are processed through dedicated phosphoproteomics search workflows (Proteome Discoverer with PTM-RS scoring, Mascot Delta CN, or Byonic) with FDR-controlled validation. Each phosphosite assignment is manually verified against diagnostic fragment ions and orthogonal fragmentation evidence.
- 6
Reporting & Regulatory Support
We deliver a comprehensive CMC-ready report including annotated MS/MS spectra for each identified phosphosite with localization scores, relative or absolute stoichiometry, method parameters, and functional relevance assessment — structured to support regulatory documentation and query responses.
Case Study: LC-MS/MS Identification of Phosphorylation on a Glycine-Serine Linker in a Bispecific Fusion Protein
In a published investigation by Tyshchuk et al. (2017) at Roche Innovation Center, an integrated LC-MS/MS strategy was employed to characterize an unexpected +79 Da modification on a recombinant bispecific IgG-based fusion protein expressed in HEK and CHO cells. The modification was initially detected by ultra-high-resolution ESI-QTOF intact mass analysis as a prominent product variant at approximately 11% abundance in HEK-expressed material — raising immediate questions about product heterogeneity and the suitability of the expression host for clinical manufacturing.
Key findings:
- Host cell dependency: 11.3% phosphorylation in HEK vs 0.4% in CHO — a 28-fold difference demonstrating that expression host selection directly impacts phosphorylation levels
- Orthogonal MS confirmation: CID produced the characteristic −98 Da neutral loss (H₃PO₄) confirming phosphorylation; EThcD unambiguously localized the site to the C-terminal serine of Linker I
- Multi-enzyme digestion: Thermolysin digestion with HCD distinguished between two identical GS-linkers in the fusion construct, pinning the modification exclusively to Linker I
- Biochemical validation: Alkaline phosphatase treatment completely removed the modification; synthetic phosphopeptide spiking with co-elution verified correct sequence assignment
- Structural insight: The (G₄S)₂ linker creates a CK2 kinase consensus motif (SSSGSSSG), demonstrating how common engineering elements can inadvertently introduce phosphorylation hotspots in fusion protein design
Implication for biologic development: Glycine-serine linkers — ubiquitous in fusion proteins, bispecific antibodies, and CAR-T constructs — can introduce unintended phosphorylation sites that create product heterogeneity. Comprehensive phosphorylation characterization during early-stage development enables informed assessment of cell line selection, molecular design, and manufacturing strategy before costly late-stage surprises.
Source: [1]
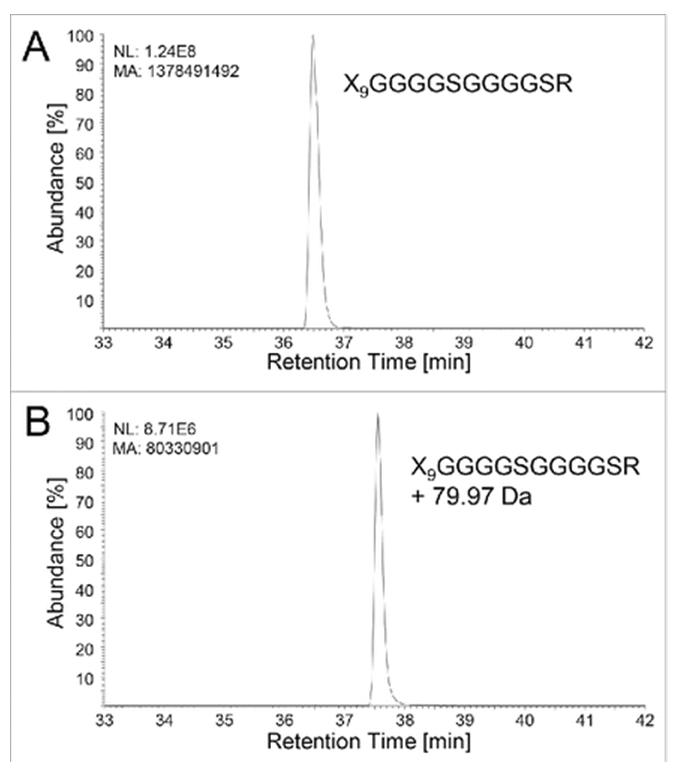
Figure from Tyshchuk et al. (2017) showing intact mass analysis and LC-MS/MS identification of phosphorylation on a glycine-serine linker in an IgG-based fusion protein (CC BY 4.0)
Frequently Asked Questions About Phosphorylation Analysis
What is the minimum sample amount required for phosphorylation site mapping?
For standard phosphosite discovery mapping, 200–500 μg of purified protein is recommended to compensate for the typically low and substoichiometric nature of phosphorylation. For targeted phosphosite quantitation using PRM, 100–200 μg may be sufficient. We assess each project individually and can optimize enrichment strategies for limited sample amounts.
How do you distinguish between phosphorylation and other modifications with similar mass shifts?
Phosphorylation (+79.966 Da on Ser/Thr/Tyr) can be distinguished from sulfation (+79.957 Da) by high-resolution accurate mass measurement on Orbitrap platforms (mass accuracy <3 ppm). Additionally, phosphorylation produces a characteristic neutral loss of phosphoric acid (H₃PO₄, −98 Da) under HCD fragmentation, while sulfation does not. ETD/EThcD fragmentation provides further orthogonal confirmation by retaining the phosphate group on the peptide backbone during electron-driven dissociation.
Can you quantify phosphorylation stoichiometry at individual sites?
Yes. We offer multiple quantitation strategies depending on your project requirements: (1) label-free quantitation by extracted ion chromatogram (XIC) peak area comparison between phosphorylated and non-phosphorylated peptide pairs; (2) targeted PRM quantitation with stable isotope-labeled synthetic phosphopeptide internal standards for absolute stoichiometry determination; and (3) SILAC or tandem mass tag (TMT) labeling for multiplexed relative quantitation across experimental conditions.
How do you prevent phosphate loss during sample preparation and MS analysis?
We incorporate several safeguards: (1) all sample preparation steps are performed with phosphatase inhibitor cocktails at controlled temperatures; (2) during MS acquisition, we use gentle ionization conditions and deploy ETD/EThcD fragmentation which preserves the labile phosphate group; (3) we include known phosphopeptide controls to monitor phosphate retention throughout the workflow; (4) alkaline phosphatase treatment is available as an orthogonal confirmation step when ambiguous assignments are encountered.
At what development stage should I consider phosphorylation characterization?
Phosphorylation characterization is valuable at multiple stages: (1) during clone selection — to identify problematic phosphosites before process development investments; (2) during process development — to monitor phosphorylation level changes in response to culture conditions; (3) during formulation development — to assess phosphorylation impact on stability; and (4) during biosimilar comparability exercises — as part of the analytical similarity assessment package. Early characterization is strongly recommended, as unexpected phosphorylation variants discovered late in development can raise regulatory questions that delay submission timelines.




Ready to Characterize Your Biologic's Phosphorylation Status?
Request Your Custom Phosphorylation Analysis PlanPartner with our experts to design a modality-specific phosphorylation characterization strategy that delivers the analytical depth and regulatory confidence needed to advance your biologic development program.
For Research Use Only. Not for diagnostic procedures.
