Why characterize protein primary structure
The molecular structure of therapeutic proteins is complex and diverse, and their properties vary widely. Therefore, specific analytical methods must be developed for various therapeutic protein molecules. The primary structure of a therapeutic protein usually refers to its amino acid sequence, which contains important structural, characteristic and genetic information. And it is the most fundamental form of describing the subcellular localization, structure and function of the protein, with potential impact on the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of its drug product. Therefore, various mass spectrometry-based analytical techniques are often required to characterize the primary structure of therapeutic proteins at multiple levels.
Mass spectrometry-based protein analysis techniques
Mass spectrometry-based protein characterization techniques
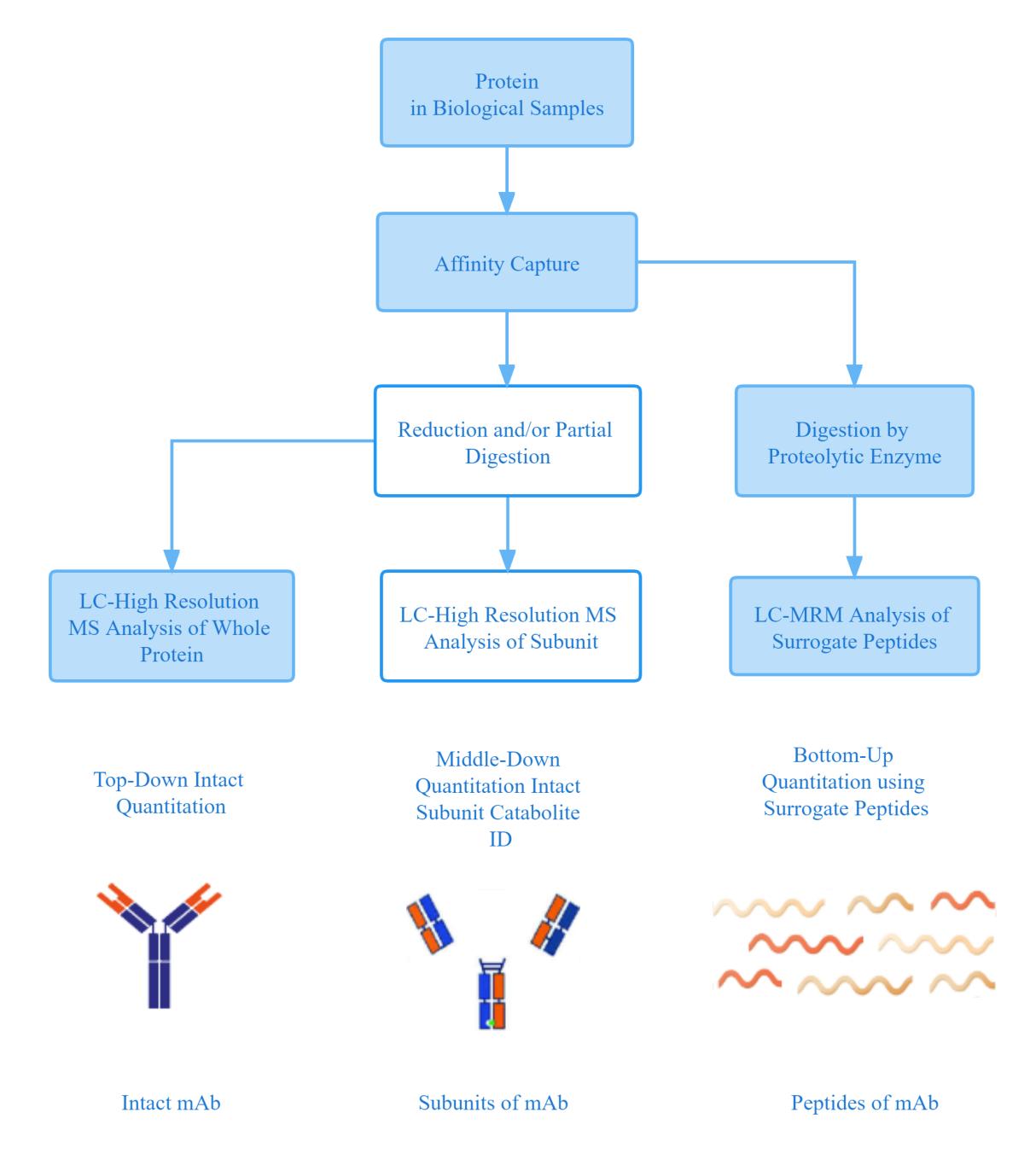
Intact Mass Analysis-liquid mass spectrometry
The first step in protein structure characterization is the mass spectrometry analysis of the complete molecule (Intact). Through this step, the mass spectrum of the entire protein molecule is obtained, and then the basic components, major modifications and glycosylation modifications of the protein are confirmed.
The resolution, precision of the mass spectrometer and sample quality directly affect the accuracy of mass spectrometry analysis. The higher the resolution, the smaller the mass difference that can be discerned.
Intact Protein Analysis
However, for large molecules such as monoclonal antibodies (mAb) and Fc-Fusion, the isotopic abundance ratio of the mass spectrometer is too wide (~25 Da), so the heterogeneity due to deamidation (+1 Da), oxidation (+16 Da), dehydration (-18 Da), etc. cannot be resolved even with isotopic resolution. When analyzing intact proteins by mass spectrometry, either ESI or MALDI ionization are typically used, with ESI producing highly multiply charged ions and MALDI mainly producing ions with small amounts of charge. Therefore, when MALDI is used to analyze protein macromolecules, a mass analyzer with a wide m/z range is required, so it is usually coupled with a TOF mass analyzer, which has an almost unlimited m/z range. Although MALDI-TOF is commonly used for mass spectrometry of large molecules such as mAb and Fc-Fusion, its mass accuracy and resolution for large molecules are poor, so ESI is mostly chosen for the heterogeneity characterization of mAb and related products.In addition, ESI is more easily coupled to liquid chromatography, which allows for LC-MS analysis.
Of all mass spectrometry instruments, Fourier transform-ion cyclotron resonance (FT-ICR) has the highest resolution and mass accuracy and is well suited for top-down analysis. However, FT-ICR instruments are expensive and uncommon in biopharmaceutical laboratories. With a wide m/z range, low ppm mass accuracy, and high resolution, ESI-TOF and ESI-QTOF are the instruments of choice for intact protein profiling. In addition, the Orbitrap mass analyzer is also well suited for the analysis of intact proteins, which also provide high resolution and low ppm mass accuracy.
Long peptides-Liquid phase mass spectrometry analysis
The Long peptides (Middle-up) Liquid phase mass spectrometry analysis is a method for reanalysis of proteins after cleavage into large fragments, specifically: reduction of disulfide bonds of the heavy chain (HC) and light chain (LC) followed by LC-MS analysis. For example, for antibody-related products, Fab or (Fab')2 and Fc fragments are formed by limited protein hydrolysis of the hinge region of HC under non-denaturing conditions. A bacterial cysteine protease specifically cleaves the IgG hinge structural domain. This method has the advantages of rapid, informative and low material consumption, and allows accurate site-by-site analysis of N-glycosylation modifications, so it can more effectively monitor the levels of rockulose and galactosylation on Fc, and also resolve and characterize C-terminal lysine cleavage, N-terminal pyroglutamylation, oxidation, truncation and uncleared various charge and size heterogeneities such as signal peptides.
Gas chromatography mass spectrometry
Gas chromatography mass spectrometry analysis of intact biomolecules is called top-down mass spectrometry, and gas-phase fragment analysis of long proteolysis produced by reduction or limited proteolysis is called middle-down. These methods can be used for sequence confirmation of terminal regions and variable domains, major modifications Identification and localization and characterization of major glycoforms. However, protein fragmentation generates a large number of highly charged fragment ions, thus requiring a high-resolution mass spectrometer. Larger molecules require higher resolution, so top-down analysis of large biomolecules remains challenging and has limited success.In contrast, the Middle-down method may be preferable, using which the protein is reduced to a smaller molecular weight fragment and then the desired resolution can be achieved with QTOF and Orbitrap instruments. In top-down/middle-down mass spectrometry analysis of intact proteins, electron transfer dissociation (ETD) and electron capture dissociation (ECD) fragments are commonly used, which provide broader sequence coverage than collision-induced dissociation (CID) and infrared multiphoton dissociation (IRMPD). Moreover, the main advantage of ETD and ECD is that they preserve unstable post-translational modifications (e.g., glycosylation) of intact proteins and can also cleave disulfide bonds. In addition, the novel high-energy inducible cleavage technology (HCD) cleaves proteins more efficiently than conventional CIDs and can also identify methionine oxidation sites. Although FT-ICR has the highest resolution, the usual method of ECD coupled with FT-ICR is only used for Top-down analysis and is not very widely used. Combining ETD with Orbitrap technology or high-resolution Q-TOF can make Top-down/middle-down analysis easier, and combining ETD with LTQ Orbitrap can further improve the coverage of specific fragment ion sequences. In addition, combining ETD or ECD with high-resolution Middle-down methods also allows fragmentation of regions protected by disulfide bonds and thus has important applications.
Peptide analysis
Peptide analysis (Bottom-up) refers to the analysis of peptide mixtures after enzymatic digestion of proteins. First, the protein is denatured, reduced, and alkylated by Cys residues, and then digested with trypsin or endoprotease (eg, Lys-C, Asp-N, or Glu-C). Then, the samples were separated using LC. Finally, the resulting proteolytic mixture was analyzed by MALDI-MS or ESI-MS. Tandem mass spectrometry is often used to sequence peptides to locate glycosylation sites, disulfide bonds, and other PTMs localization.
References
- A. Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer and S. Sanglier-Cianferani, Analytical chemistry, 2013, 85, 715-736.
- A. Beck, H. Diemer, D. Ayoub, F. Debaene, E. Wagner-Rousset, C. Carapito, A. Van Dorsselaer and S. Sanglier-Cianférani, TrAC Trends in Analytical Chemistry, 2013, 48, 81-95.
- A. Beck, M.-C. Bussat, N. Zorn, V. Robillard, C. Klinguer-Hamour, S. Chenu, L. Goetsch, N. Corvaïa, A. Van Dorsselaer and J.-F. Haeuw, Journal of chromatography B, 2005, 819, 203-218.
- P. Boomathi Pandeswaria, Varatharajan Sabareesh RSC Adv., 2019,9, 313-344.
Related Service
Protein Molecular Weight Determination
ESI-Q TOF-MS Intact Mass Analysis
MALDI-TOF-MS Intact Mass Analysis
Protein Modifications Analysis





