Characterization of HCPs
The production of therapeutic protein drugs may generate a variety of HCPs that may adversely affect the process and product performance, so methods that can identify and monitor all HCP components need to be developed to support the assessment and control of HCP-related risks in therapeutic protein products.
Traditional methods of HCP characterization and their challenges
Typically, the most used HCP assay in the production of therapeutic proteins is the enzyme-linked immunosorbent assay (ELISA). This method allows direct quantitative analysis of the overall abundance of HCP using polyclonal antibodies. However, it is generally not possible to rapidly quantify individual HCP fractions using this method and may not detect less immunogenic or non-immunogenic HCP.
A variety of complementary assays have been developed for monitoring HCP, including 1D/2D-PAGE, mass spectrometry (MS)-based analytical techniques, and others. Among them, liquid chromatography-tandem mass spectrometry (LC-MS/MS) can simultaneously identify and quantitatively analyze HCP impurities, and is the main orthogonal complementary analysis method of ELISA.
However, the biggest challenge in the application of MS-based methods is that the HCP concentration is usually 6 or more orders of magnitude lower than the target protein, and the mass spectrometer itself lacks the ability to identify and detect low concentrations of HCP from high concentrations of target proteins. To overcome the large dynamic concentration range between low ppm levels of HCP and high concentrations of therapeutic proteins, two strategies are generally available: one strategy is to add additional separation methods such as 2D-LC and/or ion mobility prior to mass spectrometry analysis to remove peptides that can co-elute with the target product, and to supplement this with data-dependent or data-independent acquisition to improve separation efficiency. However, the cycle time of 2D-LC can be long and the method is not sensitive enough to low levels of HCP less than 10 ppm. Another strategy focuses on the sample preparation process by removing the target product from the sample using affinity purification, limited digestion, or by capturing and enriching HCP using polyclonal antibodies. The most efficient method is to enrich HCPs by molecular weight cut-off (MWCO) ultrafiltration, followed by identification and analysis of the enriched HCPs.
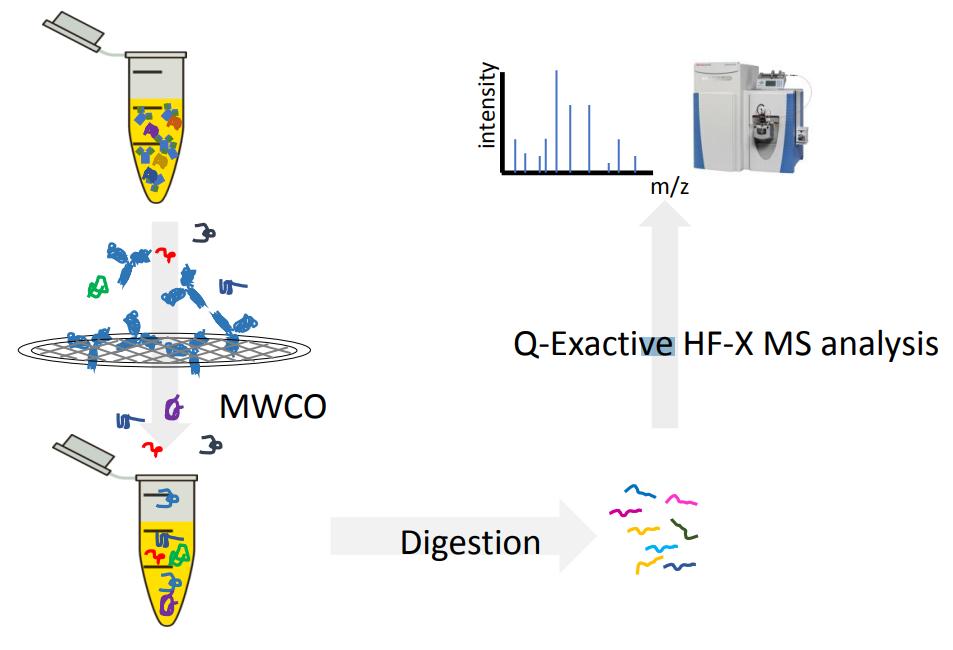
The HCP characterization technique based on MWCO[9]
Efficient MWCO-based HCP characterization technique
The MWCO-based HCP characterization technique consists of the following steps: first, the sample is treated with surfactant to release the interaction between the target protein and the HCP, and then most of the target protein is removed by molecular weight cut-off (MWCO) ultrafiltration, while the HCP in the sample is enriched to reduce the concentration difference between the HCP and the target protein. This strategy can remove most of the target proteins, significantly narrow the dynamic concentration range between HCP and target proteins, and effectively improve the sensitivity of detection of low abundance HCP.
Sample preparation and protein digestion
The samples were treated with MWCO by drying the target protein samples containing HCP and dissolving them in Tris HCl dissociation buffer at pH 8.0 containing sodium deoxycholate (SDC) and sodium lauryl sarcosinate (SLS). The samples were then flushed by ultrafiltration to remove the target protein, followed by reduction and alkylation with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and chloroacetamide (CAA). The alkylated protein samples were diluted with Tris HCl at pH 8.0 and digested with trypsin. The digested peptides were acidified with trifluoroacetic acid (TFA), and ethyl acetate was added to the digestion solution, mixed and shaken to separate the phases to form a biphasic system: the aqueous phase containing the peptides and the organic phase containing SDC and SLS. The aqueous phase was collected and dried, and the dried polypeptide mixture was resuspended and desalted in TFA solution. Finally, LC-MS/MS analysis and parallel reaction monitoring (PRM) proteomic analysis were performed on the desalted samples.
For control samples containing only the protein of interest, proceed directly to protein digestion. Samples were dried, resuspended and reduced in denaturing/reducing buffer containing urea and dithiothreitol (DTT), alkylated with iodoacetamide, and digested with trypsin. The resulting peptide mixture was acidified and diluted with TFA. Finally, LC-MS/MS and PRM analysis were performed directly.
LC-MS/MS analysis
The MWCO desalted peptide mixture was dried, resuspended in formic acid (FA) solution, and injected into an HPLC system coupled to a mass spectrometer. Peptides were separated on a C18 column, and the mobile phase buffer was aqueous FA (buffer A). The elution buffer was FA acetonitrile (ACN) solution (buffer B), and the peptide was eluted with a linear gradient in buffer B. The mass spectrometer was run in a data-dependent acquisition mode, followed by proteomic analysis of the resulting MS data.
PRM proteomic analysis
PRM is a targeted proteomics technology based on high-resolution, high-precision mass spectrometry, which can selectively detect target proteins and target peptides, so as to achieve absolute quantification of target proteins/peptides.
Data analysis
Search analysis was performed from the UniprotKB murine protein database by proteomic data analysis. The search criteria included static aminomethylation of cysteine (+57.0214 Da), oxidation of methionine residues (+15.9949 Da) and acetylation of the N-terminus of the protein (+42.011 Da) and other variable modifications. A maximum of two cleavage deletions were allowed by searching against the database after trypsin digestion, setting the false discovery rate for proteins and peptides at 0.01. Finally, the PRM data were manually curated with SKYLINE targeted proteomics software to identify HCPs using at least two specific peptides.
Overall, this strategy is a simple and efficient HCP characterization strategy that combines one-step MWCO and Shotgun proteomic analysis to identify HCPs in therapeutic protein drug products. Using this strategy removes most of the protein of interest, which greatly improves the detection of low-abundance HCPs. Therefore, the method is very sensitive, can detect HCPs with concentrations as low as 1 ppm, and can detect a relatively complete range of HCPs with high repeatability. However, the detection range of this method for HCP is also affected by the pore size of the MWCO filter. Therefore, to more fully characterize HCPs, multiple orthogonal and complementary approaches are required.
Conclusion
Cell culture-based biopharmaceutical processes generate a large number of HCPs, many of which may affect product properties and interactions with the product during production and storage, and even affect the immunogenicity, bioactivity, and safety of the product. Therefore, characterization and monitoring of HCPs is an important consideration in process development and production.
Due to the wide variety of HCPs and the wide range of their molecular weights and abundances, many of which differ from the target protein molecules by several orders of magnitude, it is difficult to perform comprehensive and accurate standards using traditional detection methods. Removal of target protein interference and enrichment of HCP by MWCO can overcome this problem, and combined with Shotgun proteomics analysis technology, the sensitivity, accuracy and reproducibility of the assay can be greatly improved. However, this method has limitations in the detection of HCP with large molecular weights and needs to be further optimized or combined with other orthogonal or complementary methods.
With the continuous progress of HCP characterization technology, the utility of proteomic databases and the available genomic information and algorithms will continue to develop, and the detection of HCP will become more and more accurate in the future, and it is expected that the immunogenicity, biological activity, safety of HCP and its interaction with the target protein will be predicted based on these data. With this critical information, standardized production and quantitative risk assessment can be better guided, process control space design can be supported, process change/improvement risks can be assessed, comparability studies can be performed, and process optimization can be used to address various situations during the product life cycle to improve the quality and stability of the final product.
References
- J. S. Bee, L. Tie, D. Johnson, M. N. Dimitrova, K. C. Jusino and C. D. Afdahl, Biotechnology progress, 2015, 31, 1360-1369.
- J. Y. Kim, Y. G. Kim, J. Y. Baik, E. J. Joo, Y. H. Kim and G. M. Lee, Biotechnology progress, 2010, 26, 246-251.
- K. Champion, H. Madden, J. Dougherty and E. Shacter, BioProcess Int, 2005, 3, 52-57.
- K. D. Ratanji, J. P. Derrick, R. J. Dearman and I. Kimber, Journal of immunotoxicology, 2014, 11, 99-109.
- K. J. Clark, F. W. Chaplin and S. W. Harcum, Biotechnology progress, 2004, 20, 1888-1892.
- K. Lintern, M. Pathak, C. M. Smales, K. Howland, A. Rathore and D. G. Bracewell, Journal of chromatography. A, 2016, 1461, 70-77.
- M. E. Cromwell, E. Hilario and F. Jacobson, The AAPS journal, 2006, 8, E572-E579.
- M. Jones, N. Palackal, F. Wang, G. Gaza‐Bulseco, K. Hurkmans, Y. Zhao, C. Chitikila, S. Clavier, S. Liu and E. Menesale, Biotechnology and bioengineering, 2021, 118, 2870-2885.
- I.-H. Chen, H. Xiao, T. Daly and N. Li, Analytical chemistry, 2020, 92, 3751-3757.
Related Service
Related Resource
What are Host Cell Proteins and Its Effect on Biopharmaceutical Development





