When mass spectrometry is employed to characterize structural variants or modifications of proteins, the measurement of the whole molecule can offer a general profile of the protein but fails to locate the specific variants or modifications. In other words, the measurement of the whole molecule does not provide any structure-resolving information. To obtain this, the protein has to be cleaved into smaller fragments prior to mass analysis. This cleavage process can be conducted either in solution or in the gaseous phase. When fragments are mainly generated in solution, the mass spectrometer is used to analyze each fragment while the molecule is reconstructed based on the information derived from these fragments, a method known as the 'bottom-up' approach. Conversely, when fragments are mainly formed in the gaseous phase within the mass spectrometer, the mass spectrometer is used to directly analyze the whole molecule, a method referred to as the 'top-down' approach.
Protein characterization methodologies utilizing mass spectrometry primarily involve three levels – top-down, middle-up, and bottom-up (involving peptide and glycan fragment ions). The bottom-up approach stands as the predominant structural characterization method, adeptly accomplishing both the resolution of the protein sequence and the identification of modification sites on the amino acid residues.
 Image source thermofisher.com
Image source thermofisher.comStructural Characterization of Bottom-Up
Bottom-up methodology is the most commonly used approach to confirm protein sequences or to characterize modifications at the residue level. In a bottom-up approach, proteins are digested into smaller peptides by protease, followed by analysis using liquid chromatography-mass spectrometry (LC/MS), or more frequently, tandem mass spectrometry (LC/MS/MS). Coupling protein hydrolysis with LC/MS/MS provides high-structural resolution, though it is typically the most material and time-consuming, as well as labor-intensive stage in mass spectrometric analysis. False positives during digestion, including the loss of labile modifications like succinimides and amino acid rearrangements, could lead to erroneous conclusions; hence careful handling is required. The bottom-up approach is widely utilized in confirming protein sequences, characterizing modifications during translation, processing, and storage, as well as describing disulfide bond connections.
Select Service
Protein Hydrolysis
Typically, monoclonal antibodies (mAbs) are first reduced and alkylated under denaturing conditions, after which trypsin or Lys-C is utilized for protein digestion. Other less frequently used proteases include Glu-C (V8) and Asp-N. Occasionally, chemical cleavage methods, such as Cyanogen Bromide (CNBr) digestion, are also employed.
These methodologies generally cleave proteins into peptides of an appropriate size, which can be effectively fragmented in MS/MS experiments. It's important to note, on most commercially available instruments, large peptides (>4 kDa) are challenging to characterize through MS/MS, whereas extremely small peptides (2-3 residues) are often lost due to poor retention on reversed-phase columns.
LC/MS and LC/MS/MS
In the early stages, MALDI-TOF instruments were widely employed to determine the masses of peptides generated from the enzymatic digestion of monoclonal antibodies (mAbs) and other proteins, serving for structural verification and isomer/ modification analysis. Compared to ESI-MS, MALDI-TOF offers advantages of user-friendliness and less consumption of supplies and time. However, due to the convenience of LC coupling and the superior tandem mass spectral quality required for peptide identification, most current bottom-up experiments conducted on mAbs are performed on ESI instruments.
Challenges often arise during the protein hydrolysis of monoclonal antibodies (mAbs) in LC/MS/MS experiments. A significant amount of protein is typically needed to achieve a successful digestion. As long as complete digestion is realized, sensitivity in MS detection is not usually an issue, due to significant advancements in contemporary instrumentation. Therefore, most LC/MS/MS experiments are performed on narrow-bore reversed-phase ultra-high performance liquid chromatography (RP-UPLC) columns to attain optimal chromatographic resolution.
Disulfide Bridges Determination
The elucidation of disulfide bridge formation within a protein molecule requires the protein to be digested while disulfide bonds remain integral. Monoclonal antibodies (mAbs) prove to be considerably stable and resilient to digestion, under standard conditions, thereby making their disulfide bridging characterization a challenging endeavor. The key to successful digestion lies within the denaturation of the mAb under harsh conditions, such as using 6M Guanidine HCl at elevated temperatures. It is crucial that an alkylating agent, such as iodoacetic acid (IAA), iodoacetamide (IAM), or N-Ethylmaleimide (NEM), be present during the denaturation process to impede any free thiol group within the mAb, thereby preventing disulfide scrambling. NEM is usually the preferred choice due to its activity at a lower pH. Prior to digestion, it is necessary to dilute the denaturant to preserve enzyme activity. Digestion typically involves the use of trypsin or Lys-C. Following this, a portion of the sample will undergo further disulfide reduction using DTT or TCEP, and a comparison is made between LC/MS/MS peptide maps of reduced and non-reduced peptides to ascertain the disulfide linkage. Disulfide bonds close to the hinge region are generally detected by partial reduction using TCEP in weak acid conditions in the presence of NEM or M-Biotin, followed by Edman sequencing.
Select Service
The Determination of Disulfide Bonds in mAbs with LC/MS Analysis in Negative Ion Mode Another useful approach for ascertaining disulfide bonds in monoclonal antibodies (mAbs) involves analyzing non-reduced digestible material through LC/MS in negative ion mode. In this mode, disulfide bonds are ordinarily effectively cleaved, revealing chains connected via disulfide linkages. This method offers a type of gas-phase disulfide bond reduction.
Top-Down Structural Characterization
Despite the bounty of information provided by bottom-up approaches, they require substantial manpower and encounter issues such as the necessity for extensive sample consumption, possible introduction of artifacts during digestion, and significant time involvement for sample preparation. In instances where sample quantity or concentration is limited - such as minor components acquired from chromatographic separation - multiple rounds of collection and concentration are required in order to accrue enough material for successful digestion. In such scenarios, top-down and middle-down emerge as remarkably rapid and convenient alternatives for delivering valuable sequence information.
Top-Down mass spectrometry refers to the analysis of intact molecules without prior enzymatic or chemical digestion in solution, gaining structural information through the examination of fragmentation patterns within the mass spectrometer. The top-down approach has successfully enabled rapid characterization of small to medium-sized proteins. Owing to the substantial quantity of highly charged fragment ions yielded, high-resolution mass spectrometers are typically required for top-down analysis. The size of the protein amenable for top-down mass spectrometry analysis is generally determined through the resolving power of the instrument. For instance, Quadrupole Time of Flight (Q-TOF) instruments with a resolution of 10,000 should provide extensive structural information for proteins up to 10,000 Da in mass. For larger proteins, instruments with higher resolution, such as Fourier Transform Ion Cyclotron Resonance mass spectrometry (FT-ICR MS) or Orbitrap, are the preferred choice.
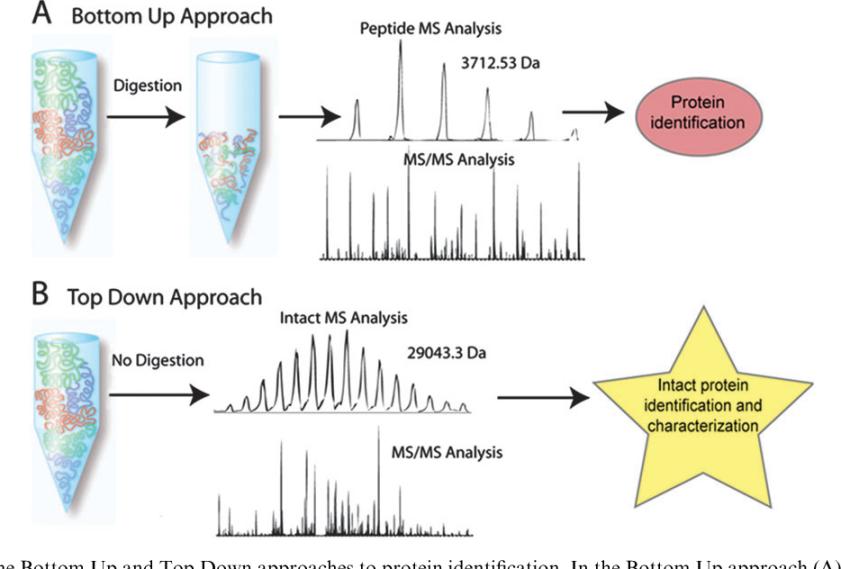 Bottom Up and Top Down approaches (John F. Kellie et al,. 2010)
Bottom Up and Top Down approaches (John F. Kellie et al,. 2010)Middle-Up Protein Characterization
Whole monoclonal antibody (mAb) mass measurement offers a comprehensive overview of a protein; however, it lacks the power of resolution necessary for characterizing structural features. To gain structural information, proteins must be dissected into smaller units prior to mass analysis. An alternative approach, known as the 'Middle-Up' method, involves partitioning proteins (or mAbs) into several larger segments before performing mass spectrometry. A practical strategy to disintegrate mAbs into several large segments utilizes disulfide reduction to generate light and heavy chains. Analyzing the mass of these individual chains can confirm their respective structures, or pinpoint variants or modifications within each separate chain.
Reduction of all disulfide bonds can be performed under denaturing conditions. Selective reduction of interchain disulfide bonds, while leaving intrachain disulfide bonds intact, can be performed without denaturants. This straightforward reduction method has been used for the confirmation of mAb structures, characterization of antibody conformations, identification of distinctive modifications on light and heavy chains, and examination of glycosylation structures on the heavy chain.
Another popular Middle-Up approach involves proteolytic treatment of mAbs under native conditions using enzymes such as papain or pepsin. For many IgG molecules, these limited proteolysis cleavage sites tend to be near the hinge region, generating Fab, F(ab)2, and Fc fragments. However, IgG2 molecules resist enzymatic digestion under native conditions. The fragments produced by these proteolytic treatments yield smaller segments upon reduction. The mass analysis of these fragments offers more detailed structural information than mass measurements of the intact mAb. Similar to the reduction approach, limited proteolysis approaches have been utilised for confirming mAb structure, identifying post-translational or chemical modifications, and characterising antibody architecture.
Middle-Down Structural Characterization
Middle-down characterization might serve as a valuable alternative to address the limited structural resolution inherent in top-down methodologies. In middle-down approaches, proteins are first dissected into several large fragments via either reduction of disulfide bonds or limited proteolysis as an intermediary step prior to introduction to the mass spectrometer for MS/MS analysis. As opposed to top-down, middle-down has the potential to enhance structural resolution in cases with limited sample preparation. Given that the reduction of disulfide bonds used in middle mass experiments involves a chemical reaction where reagents can be used in excess, it does not impose sample concentration demands, unlike enzymatic processes that typically do not employ a large quantity of enzyme due to cost considerations. Thus, for dilute or minute samples, middle-down experiments offer a more advantageous standing than top-down.
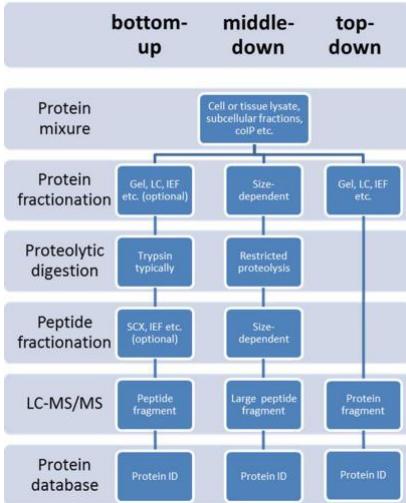 bottom-up vs top-down vs middle down. (Abeer S. Alhendi 2020)
bottom-up vs top-down vs middle down. (Abeer S. Alhendi 2020)