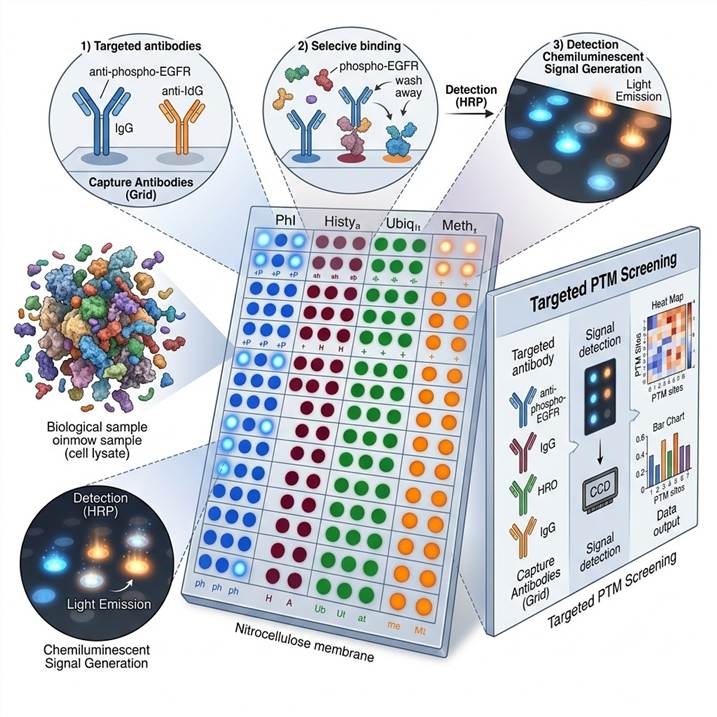
Why PTM Antibody Array Profiling
Antibody-based PTM arrays occupy a distinct and essential niche in the PTM analysis ecosystem. While mass spectrometry-based discovery provides the broadest coverage for identifying novel modification sites, antibody arrays deliver unique advantages for targeted, hypothesis-driven PTM analysis across multiple biological samples and experimental conditions.
Complementary to Mass Spectrometry-Based PTM Discovery
MS-based PTM proteomics excels at unbiased discovery — identifying thousands of modification sites without prior knowledge of which proteins or residues are modified. Antibody arrays, by contrast, provide targeted, hypothesis-driven analysis of predefined PTM panels with established antibodies validated for specificity and sensitivity. This makes antibody arrays the ideal platform for: validation of MS discovery hits across larger sample cohorts, time-course and dose-response experiments requiring many samples, screening applications where rapid turnaround is prioritized over discovery breadth, and research settings without direct access to specialized MS instrumentation.
Pathway-Level Signaling Network Analysis
PTM antibody arrays are particularly powerful for phospho-signaling network analysis. Phospho-kinase arrays, for example, simultaneously profile the activation status of 30–50+ key kinases and signaling nodes, providing a systems-level snapshot of cellular signaling pathway activity from a single sample. This makes them invaluable for understanding drug mechanism of action, identifying signaling pathway rewiring in disease, and profiling the functional consequences of genetic perturbations. Similarly, histone modification antibody arrays enable comprehensive profiling of the histone code — the combinatorial pattern of methylation, acetylation, and phosphorylation marks on histone proteins that governs chromatin state and gene expression.
Rapid, High-Throughput PTM Screening
Compared to MS-based workflows that require days to weeks of sample preparation, data acquisition, and bioinformatics analysis, antibody arrays provide results within a single day from sample preparation to data readout. This speed advantage, combined with the ability to process dozens of samples in parallel, makes antibody arrays the method of choice for screening applications, time-course studies, and multi-condition comparisons where the priority is rapid, reliable PTM profiling rather than comprehensive discovery. For broader, integrated PTM analysis combining discovery and validation, our Global PTM Profiling and MS-based platforms provide the complementary discovery capability to identify novel targets for subsequent antibody array validation.
Our PTM Antibody Array Platform
Our PTM antibody array platform deploys validated commercial and custom antibody array formats optimized for different PTM classes, sample types, and throughput requirements. Each format is selected based on your specific research question and sample availability.
Membrane-Based Antibody Arrays
Our standard membrane-based antibody array format uses nitrocellulose membranes printed with duplicate spots of capture antibodies specific to PTM-modified and total protein targets. Samples are incubated with the membrane, and bound target proteins are detected using a cocktail of detection antibodies followed by chemiluminescent or fluorescent readout. This format is ideal for profiling 20–100 targets across multiple samples with minimal sample input (200–500 µg total protein per membrane). The membrane format supports phospho-kinase arrays, phospho-RTK arrays, apoptosis arrays, and custom PTM panels.
Glass Slide and Bead-Based Multiplex Arrays
For higher-density multiplexing (100–1,000+ targets per sample), we offer glass slide-based antibody microarray and bead-based multiplex assay formats. Glass slide arrays enable simultaneous profiling of hundreds of PTM-specific antibodies using a sandwich or direct labeling detection format with fluorescent scanning. Bead-based multiplex assays use color-coded bead populations, each coupled to a specific PTM capture antibody, with flow cytometric readout for quantitative, high-precision detection. These formats are particularly suited for comprehensive phosphoproteome and acetylome profiling where broad coverage is needed in a targeted format.
Histone Modification Antibody Arrays
Our specialized Histone Modification Antibody Array provides comprehensive profiling of the histone code, covering methylation (mono-, di-, tri-methylation at multiple lysine and arginine residues), acetylation (H3K9ac, H3K27ac, H4K16ac, and others), phosphorylation (H3S10ph, H3S28ph), and other histone marks. Similarly, our Phospho-Signaling Antibody Array focuses on kinase activation status and phospho-signaling network analysis across multiple parallel signaling pathways. For broader PTM coverage beyond phosphorylation and histone marks, our Multiplex PTM Immunoassay Services extend antibody-based detection to acetylation, ubiquitination, SUMOylation, and other modification types.
Available PTM Antibody Array Panels
Our PTM antibody array platform offers the following major panel categories, each customizable to your specific research needs. Multiple panels can be deployed in parallel for comprehensive PTM screening.
Phospho-Kinase and Signaling Arrays
- Phospho-Kinase Array — 30–50 kinase phosphorylation site targets covering MAPK, PI3K/Akt, JAK/STAT, TGF-β, Wnt/β-catenin, and NF-κB signaling pathways. Each target is detected with phosphosite-specific antibodies, providing a comprehensive activation snapshot of major signaling networks from a single sample.
- Phospho-Receptor Tyrosine Kinase (RTK) Array — 40–50 RTK phosphorylation targets including EGFR, ErbB2/HER2, FGFR, IGFR, PDGFR, VEGFR, and EphR families, enabling rapid profiling of RTK activation patterns in cancer and developmental biology research.
- Phospho-Specific Pathway Arrays — Focused panels targeting individual signaling pathways (MAPK, PI3K/Akt/mTOR, JAK/STAT, TGF-β/Smad) with dense coverage of phosphorylation sites within each pathway for detailed pathway activation mapping.
Histone Modification Arrays
- Histone Pan-Modification Array — Comprehensive coverage of methylation (H3K4me1/2/3, H3K9me1/2/3, H3K27me1/2/3, H3K36me1/2/3, H4K20me1/2/3, H3R17me, H3R26me), acetylation (H3K9ac, H3K14ac, H3K27ac, H3K56ac, H4K5ac, H4K8ac, H4K12ac, H4K16ac), and phosphorylation (H3S10ph, H3S28ph, H2AX S139ph) marks on core and variant histones.
- Custom Histone Code Panels — Focused subsets of histone marks tailored to your epigenetic research question, with flexible panel composition to balance coverage breadth with sample throughput.
PTM and Modification-Specific Arrays
- Acetylation Array — Pan-acetyl-lysine and site-specific acetylation antibodies covering both histone and non-histone acetylation targets involved in metabolism, transcription, and cytoskeletal regulation.
- Ubiquitination and SUMOylation Arrays — Antibody panels targeting ubiquitin-modified proteins and SUMO1/2/3 conjugates, providing complementary coverage to our MS-based ubiquitin and SUMO analysis services.
- Apoptosis and Cell Signaling Arrays — Modified protein targets involved in apoptosis (cleaved caspases, Bcl-2 family), DNA damage response (γH2AX, p53 phosphorylation), and cell cycle regulation (cyclins, CDKs, CDK substrate phosphorylation).
For pathway-level validation of phosphoproteomics and PTM discovery data, our platform integrates seamlessly with our PTMs in Drug Discovery services. Downstream PTM Bioinformatics Analysis provides pathway mapping, network analysis, and integration of antibody array data with MS-based PTM datasets.
Workflow: From Sample to PTM Array Data
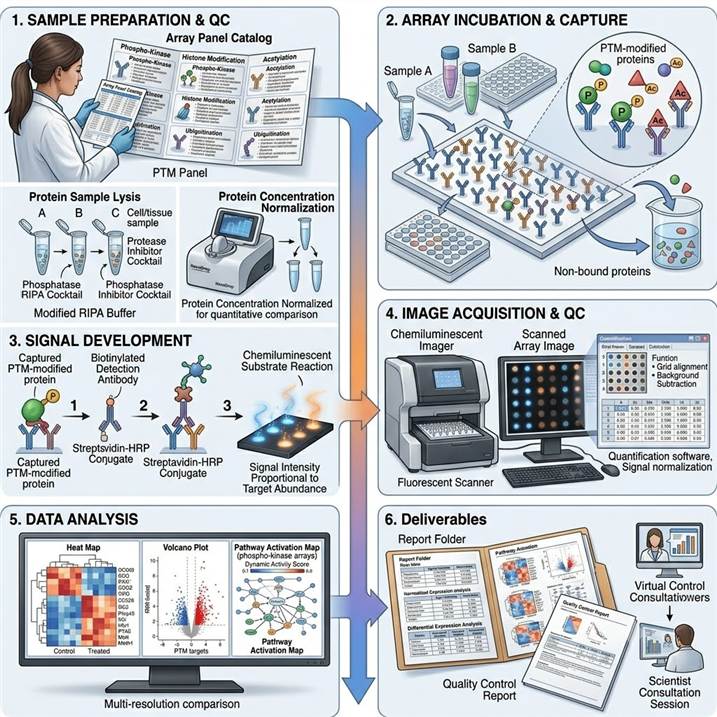
Step 1: Panel Selection and Sample Preparation
We collaborate with you to select the optimal PTM antibody array panel(s) based on your research question, PTM targets of interest, and sample type. Samples are prepared under conditions that preserve PTM integrity: cells or tissues are lysed in modified RIPA buffer containing protease and phosphatase inhibitor cocktails, and protein concentration is normalized across samples for quantitative comparison.
Step 2: Array Incubation and Target Capture
Normalized lysates are incubated with antibody array membranes, slides, or bead sets under optimized binding conditions. PTM-modified and total target proteins in the sample are captured by the immobilized antibodies. After incubation, unbound proteins are removed through stringent washing steps, and the arrays are prepared for detection antibody incubation.
Step 3: Detection and Signal Development
Captured target proteins are detected using a cocktail of biotinylated detection antibodies followed by streptavidin-HRP conjugates or fluorescent detection reagents. Chemiluminescent or fluorescent signals are developed and captured using optimized imaging systems with appropriate exposure times to ensure signal linearity across the dynamic range of each array.
Step 4: Image Acquisition and Data Extraction
Array images are acquired at high resolution using chemiluminescent imagers or fluorescent scanners. Spot intensity data are extracted using dedicated array analysis software, with background subtraction, spot quality filtering, and normalization against positive controls and reference spots. Replicate spots within each array provide technical reproducibility assessment.
Step 5: Data Analysis and Interpretation
Normalized signal intensities for each PTM target are compared across samples, with statistical analysis identifying targets with significantly altered PTM levels between conditions. For phospho-kinase arrays, pathway activation scores are calculated based on the combined phosphorylation status of multiple targets within each signaling pathway. For histone modification arrays, combinatorial mark analysis reveals coordinated changes in the histone code.
Step 6: Deliverables and Reporting
PTM antibody array data package including: raw and normalized signal intensities for each target, fold-change and statistical significance for pairwise and multi-group comparisons, heat maps and volcano plots for data visualization, pathway activation maps for signaling arrays, and a scientist consultation session for biological interpretation and integration with other PTM analysis data.
Why Choose Our PTM Antibody Array Profiling Service
Validated Antibody Panels and QC-Controlled Arrays
Every antibody used in our PTM arrays is validated for specificity, sensitivity, and cross-reactivity before incorporation into array panels. Each batch of arrays undergoes quality control testing with positive and negative control samples to ensure lot-to-lot consistency and reliable performance across projects and time points.
Integrated PTM Analysis Ecosystem
Our antibody array platform is not an isolated service — it is integrated within our comprehensive PTM analysis ecosystem that includes MS-based discovery, targeted quantification, and bioinformatics analysis. This integration means that hits from antibody array screening can be seamlessly followed up with deeper MS-based analysis of specific modification sites, or conversely, MS discovery findings can be validated across larger sample cohorts using antibody arrays — all within a single service provider.
Flexible Panel Design and Customization
While we offer extensive pre-configured panels for common PTM classes, we also provide custom array design services to incorporate your specific targets of interest. Whether you need to add custom antibodies to an existing panel, create a focused sub-panel for a specific pathway, or design a completely new array for a novel PTM target combination, our flexible manufacturing capabilities accommodate your specific requirements.
Cross-Platform Compatibility
Our antibody array data can be integrated with MS-based PTM datasets, western blot validation, ELISA, and other immunoassay results, enabling a unified analysis across orthogonal PTM detection platforms. This cross-platform compatibility ensures that your PTM profiling data can be leveraged across your entire research program regardless of the detection method used at each stage.
Case Study: Phosphoproteome Microarray Analysis of Extracellular Particles as Biomarkers for Alzheimer's Disease
In a 2024 study published in the International Journal of Molecular Sciences (MDPI), Soares Martins et al. used high-density antibody microarrays to profile phosphorylation patterns in blood-derived extracellular particles (bdEPs), demonstrating the power of antibody-based PTM arrays for biomarker discovery and clinical translation.
Background: Alzheimer's disease (AD) diagnosis currently relies on clinical assessment, cerebrospinal fluid analysis, and brain imaging — all with significant limitations in accessibility, cost, and early detection. Blood-based biomarkers would transform AD diagnosis and monitoring, but identifying sensitive and specific protein markers in peripheral samples has been challenging due to the low abundance of brain-derived proteins in circulation. Extracellular particles (including exosomes) derived from the brain cross the blood-brain barrier and can be isolated from peripheral blood, offering a window into brain pathology — but their limited protein content demands highly sensitive and multiplexed detection methods.
Approach: The team employed high-density antibody microarrays containing 1,145 pan-specific antibodies (targeting total protein levels) and 913 phosphosite-specific antibodies (targeting specific phosphorylation sites) to profile extracellular particles isolated from serum of AD patients and age-matched controls. This comprehensive antibody array platform enabled simultaneous measurement of over 2,000 protein and phosphoprotein targets from each sample, requiring no specialized mass spectrometry infrastructure. Data were analyzed using supervised learning approaches to identify discriminatory protein and phosphorylation markers, with pathway analysis providing biological context for the identified signatures.
Key Findings:
- 150 proteins showed significantly altered expression and/or phosphorylation patterns in AD patient extracellular particles compared to controls, revealing a broad dysregulation of protein phosphorylation in AD
- Differentially phosphorylated proteins included established AD-associated pathways (tau phosphorylation, amyloid processing) as well as novel candidates in synaptic function, neuroinflammation, and metabolic regulation
- The antibody microarray approach successfully detected phosphorylation changes from as little as 50 µg of extracellular particle protein per sample, demonstrating the sensitivity needed for clinical biomarker applications
- Machine learning-based classification using the phosphoproteomic microarray data achieved high discriminatory accuracy between AD and control samples, supporting the diagnostic potential of blood-derived extracellular particle phosphoproteomics
- Pathway analysis of differentially phosphorylated proteins identified dysregulation of PI3K/Akt, MAPK, and mTOR signaling pathways in AD extracellular particles, consistent with known AD pathology
Significance: This study demonstrated that high-density phosphosite-specific antibody microarrays provide the sensitivity, throughput, and reproducibility needed for clinical biomarker discovery from limited clinical specimens. The integration of 900+ phosphosite-specific antibodies into a single array format enabled comprehensive phosphoproteome profiling without MS instrumentation — establishing antibody arrays as a powerful platform for translating PTM biology into clinically actionable biomarkers. The workflow and analytical framework validated in this study are directly applicable to other disease contexts and sample types.
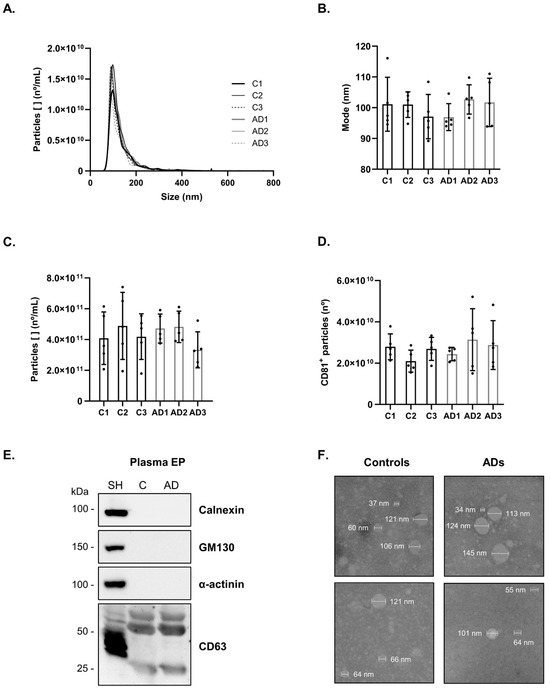
Figure 1 from Soares Martins et al. (2024). High-density antibody microarray workflow for phosphoproteomic profiling of blood-derived extracellular particles in Alzheimer's disease biomarker discovery. (CC BY 4.0)
Representative PTM Antibody Array Profiling Results
Our PTM antibody array platform delivers comprehensive data packages designed for immediate biological interpretation and publication-quality visualization of PTM signaling network changes.
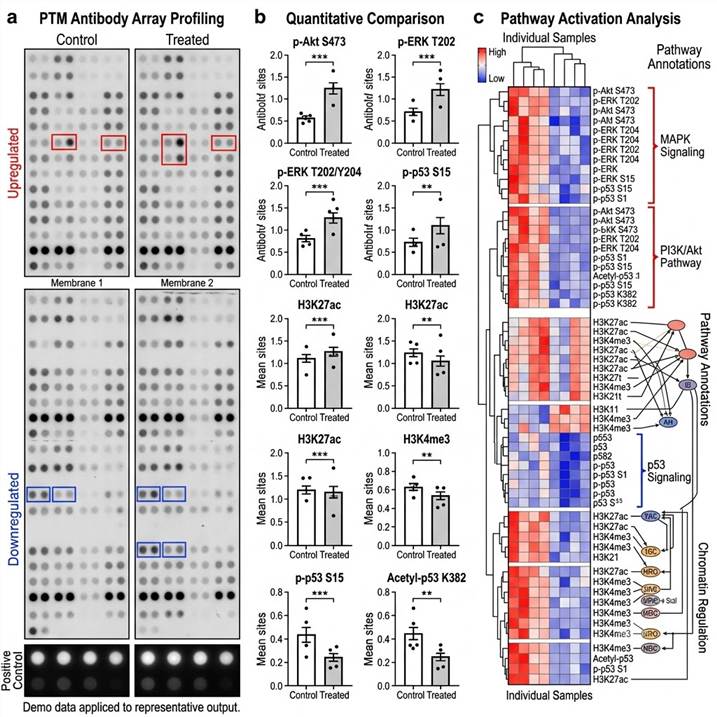
Representative data outputs from our PTM antibody array platform. Left: Array membrane images. Center: Quantitative comparison of PTM targets. Right: Pathway activation heat map and signaling network analysis.
Key deliverables included in every PTM antibody array project:
- Raw and normalized signal intensities — For each target on the array, with background subtraction, normalization to internal controls, and replicate spot quality metrics
- Differential expression analysis — Fold-change, p-value, and false discovery rate for comparisons between experimental groups, with appropriate statistical testing
- Data visualization — Publication-quality heat maps, volcano plots, scatter plots, and bar charts for data exploration and presentation
- Pathway activation analysis — For signaling arrays, integrated pathway maps showing the activation status of each signaling node and pathway-level activity scores
- Quality control report — Array performance metrics including positive control signal intensity, background uniformity, replicate spot reproducibility, and detection sensitivity for each target
Related Services
Our PTM antibody array platform is part of a comprehensive PTM analysis service portfolio spanning antibody-based and MS-based detection platforms for integrated PTM research.
- Ultra-Sensitive Modified Protein Detection — Highly sensitive immunoassay-based detection of low-abundance PTM-modified proteins using proximity-based and amplification technologies
- Bioorthogonal PTM Labeling Services — Chemical probe-based labeling and detection of specific PTM types using bioorthogonal chemistry and enrichment strategies
- PTMs in Biological Research — Integrated PTM analysis solutions for basic biology research, from signaling studies to functional characterization
- Multi-PTM Crosstalk Profiling — Integration of multiple PTM datasets for comprehensive analysis of modification crosstalk, competition, and coordinated regulation in biological systems
- PTM Crosstalk Analysis — Dedicated analysis service for investigating interactions, competition, and coordinated regulation between different PTM types on the same or interacting proteins
- Epigenetic PTM Research Services — Comprehensive epigenetic PTM analysis platform covering histone modifications, chromatin regulators, and DNA methylation-related PTMs
FAQs
What is the difference between PTM antibody arrays and mass spectrometry-based PTM analysis?
PTM antibody arrays use immobilized modification-specific or modification site-specific antibodies to capture and detect known PTM targets, providing targeted, hypothesis-driven analysis of predefined modification panels. MS-based PTM analysis uses mass spectrometry for unbiased discovery of modification sites across the entire proteome. The two approaches are complementary: MS excels at discovering novel modification sites, while antibody arrays excel at validating and screening known modifications across many samples with higher throughput and without the need for specialized MS instrumentation.
How many targets can be analyzed simultaneously in a PTM antibody array?
Our antibody array platform covers a broad range of multiplexing capacities: membrane-based arrays typically profile 20–100 targets per sample, glass slide-based microarrays can profile 100–500+ targets, and bead-based multiplex assays can reach 1,000+ targets per sample. The optimal format depends on your specific research question, sample availability, and desired throughput. We provide complimentary consultation to select the best format for your project.
What types of PTMs can be detected by antibody arrays?
Our PTM antibody arrays cover a wide range of modification types including phosphorylation (phospho-tyrosine, phospho-serine/threonine, site-specific phospho-antibodies), histone modifications (methylation, acetylation, phosphorylation, citrullination on core and variant histones), acetylation (pan-acetyl-lysine and site-specific acetyl-antibodies on histone and non-histone proteins), ubiquitination, SUMOylation, and apoptosis-specific cleaved protein targets. Custom panels can be developed for additional PTM types as needed.
What is the minimum sample amount required for PTM antibody array analysis?
Sample requirements depend on the array format and number of targets. For membrane-based arrays (20–50 targets), we typically recommend 200–500 µg of total protein per membrane. For high-density glass slide microarrays (100–500+ targets), 50–200 µg of total protein is sufficient. For bead-based multiplex assays, 25–100 µg of total protein per assay is typically adequate. Specific recommendations are provided during project planning based on your sample type and chosen array panel.
How do you ensure antibody specificity in PTM arrays?
Every antibody used in our PTM arrays undergoes rigorous validation including: peptide competition assays to confirm modification-specific recognition, cross-reactivity testing against closely related modification states and sequence motifs, western blot confirmation of target size and signal specificity, and batch-to-batch consistency testing. For phosphosite-specific antibodies, we additionally verify that signals are abolished by phosphatase treatment, confirming phosphorylation-dependent recognition.
Can PTM antibody arrays be used for quantitative comparison across multiple conditions?
Yes — quantitative comparison is one of the primary applications of PTM antibody arrays. All array formats support multi-sample comparison with appropriate normalization strategies (total protein normalization, positive control normalization, and reference sample normalization for inter-array comparisons). For time-course and dose-response experiments, we recommend processing all samples in a single batch to minimize technical variation. For large multi-batch studies, we implement bridge sample controls to enable cross-batch data integration.
How do I select between phospho-kinase array, histone modification array, and custom PTM panels?
Selection depends on your research focus. Choose phospho-kinase arrays for cell signaling pathway analysis, drug mechanism of action studies, and kinase activation profiling. Choose histone modification arrays for epigenetic research, chromatin biology, and transcriptional regulation studies. Choose custom panels when your focus is on a specific modification type (e.g., acetylation, ubiquitination) or a defined set of proteins across multiple PTM types. We provide complimentary scientific consultation to help you select or design the optimal panel for your experiment.
Can antibody array results be validated by orthogonal methods?
Yes — we recommend orthogonal validation of key findings from antibody array experiments using complementary methods. Depending on the target and modification of interest, validation can be performed using western blot with modification-specific antibodies, ELISA for quantitative single-target confirmation, or targeted MS-based methods (PRM, MRM) for site-specific validation of phosphorylation and other PTMs. Our integrated platform can perform both antibody array screening and orthogonal validation within the same service workflow.
References
- Soares Martins T, Pelech S, Ferreira M, Pinho B, Leandro K, Pereira de Almeida L, Breitling B, Hansen N, Esselmann H, Wiltfang J, da Cruz e Silva OAB, Henriques AG. Phosphoproteome Microarray Analysis of Extracellular Particles as a Tool to Explore Novel Biomarker Candidates for Alzheimer's Disease. International Journal of Molecular Sciences. 2024;25(3):1584.
- Blair JD, Hartman A, Zenk F, Wahle P, Brancati G, Dalgarno C, Treutlein B, Satija R. Phospho-seq: integrated, multi-modal profiling of intracellular protein dynamics in single cells. Nature Communications. 2025;16:1346.
- Alves J, Sligar M, Zhang H, Moyer T, Auerbach S, Guttilla IK, Cardelli JJ. Monitoring phosphorylation and acetylation of CRISPR-mediated HiBiT-tagged endogenous proteins. Scientific Reports. 2024;14:2138.
For research use only. Not for use in diagnostic procedures.