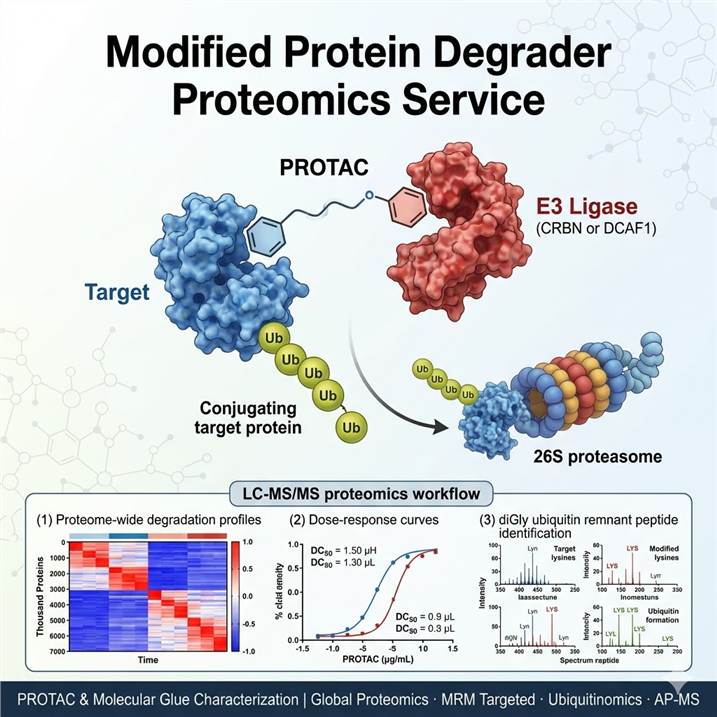
Meeting the Proteomics Challenge of Targeted Protein Degrader Development
The targeted protein degradation field has advanced rapidly from a chemical biology concept to a clinically validated therapeutic modality, with multiple PROTACs and molecular glues now in clinical trials for oncology, immunology, and beyond. However, the unique mechanism of action of degraders — catalytic, event-driven pharmacology mediated through the ubiquitin-proteasome system — imposes distinct analytical requirements that conventional pharmacokinetic/pharmacodynamic (PK/PD) assays alone cannot address. Proteomic characterization of degrader activity must simultaneously answer four interconnected questions: (1) Is the intended target degraded, and with what potency and kinetics? (2) What off-target proteins are also depleted, defining the selectivity footprint? (3) Is the degradation mechanism E3 ligase-dependent and ubiquitination-mediated? (4) What are the downstream proteomic consequences of target loss? Our integrated degrader proteomics platform deploys complementary LC-MS/MS methodologies — global proteomic profiling by DIA and TMT, targeted quantification by MRM/PRM, ubiquitin remnant diGly proteomics, and affinity purification-mass spectrometry (AP-MS) — each optimized to address specific questions across the TPD discovery and development pipeline. For targeted degradation efficacy assessment, our MRM Protein Degrader Efficacy service provides precise quantification of target protein degradation with DC50/DC90 parameter calculation.
Why Mass Spectrometry Proteomics Is Essential for TPD Development
Unlike occupancy-driven pharmacology (where drug-target binding is the primary readout), event-driven pharmacology of degraders requires direct measurement of protein loss across the entire proteome. Mass spectrometry-based proteomics is uniquely positioned to deliver this information: global discovery proteomics (DIA/TMT/Label-Free) provides unbiased, proteome-wide coverage of degradation effects with thousands of quantified proteins per experiment; targeted proteomics (MRM/PRM) delivers the analytical sensitivity and throughput needed for routine DC50 determination, dose-response curves, and time-course studies in preclinical and translational settings; ubiquitin remnant profiling directly measures the ubiquitination events that are the mechanistic prerequisite for proteasomal degradation; and AP-MS interactome analysis validates ternary complex formation and identifies E3 ligase-dependent neo-substrates. By integrating these complementary approaches, our platform provides the comprehensive proteomic characterization that differentiates successful TPD programs from those limited by selectivity issues, mechanistic uncertainty, or incomplete target engagement data.
Our Modified Protein Degrader Proteomics Service Portfolio
We offer a structured portfolio of degrader proteomics services designed to address specific characterization needs across the TPD discovery and development pipeline. The table below maps common research objectives to our recommended service modules and analytical platforms.
| Research Objective |
Recommended Service |
Key Technology |
| Proteome-wide on-target degradation and off-target selectivity profiling |
TMT-based Proteomics / Label-Free Proteomics |
DIA or TMT multiplexed quantification (10-plex to 18-plex), Orbitrap HRAM, 8,000+ proteins quantified per experiment |
| Targeted quantification of specific protein degradation with DC50 calculation |
MRM/PRM Degrader Efficacy |
QQQ MRM with isotope-labeled peptide standards (AQUA/PSAQ), scheduled MRM acquisition, DC50/DC90 curve fitting |
| Ubiquitination site mapping and E3 ligase mechanistic confirmation |
Ubiquitination Proteomics |
Anti-diGly (K-ε-GG) antibody enrichment, LC-MS/MS, ubiquitination site identification and stoichiometry |
| Target-E3 ligase ternary complex validation and neo-substrate discovery |
AP-MS Interactome Analysis |
Affinity purification with FLAG/Streptavidin pull-down, LC-MS/MS, label-free quantification, SAINT scoring |
| Degradation kinetics and half-life determination |
SILAC Degrader Kinetics |
SILAC pulse-chase labeling, heavy/light ratio tracking over time, degradation rate constant (kdeg) calculation |
| Mechanism of action deconvolution and resistance profiling |
Integrated Multi-Omics TPD Package |
Combined global proteomics + ubiquitinomics + AP-MS + phosphoproteomics, multi-modal data integration |
Each service module is available independently or can be combined into an integrated multi-parametric TPD characterization workflow. For studies requiring complementary E3 ligase activity assessment beyond degradation readouts, our PTM Enzyme Activity and Inhibitor Screening service provides targeted activity assays for ubiquitin-related enzymes and other PTM-modifying enzymes.
Key Degradation Parameters Quantified by Our Proteomics Platform
The following table summarizes the key degradation parameters routinely calculated from our proteomics data outputs, providing standardized metrics for degrader potency, selectivity, and efficacy comparisons across programs.
| Parameter |
Definition |
Quantification Method |
Typical Throughput |
| DC50 |
Degrader concentration achieving 50% target depletion |
MRM/PRM or TMT 10-point dose-response curve fitting |
10–100 samples per study |
| DC90 / DCmax |
Degrader concentration achieving 90% / maximal target depletion |
MRM/PRM at 3–5 concentrations, nonlinear regression (4PL) |
10–50 samples per study |
| Dmax (%) |
Maximum degradation level achieved (fraction remaining) |
TMT/Label-free global proteomics, MRM validation |
Included in dose-response studies |
| t1/2 (degradation half-life) |
Time required for 50% target protein loss after degrader treatment |
SILAC pulse-chase or cycloheximide chase + MRM time course |
4–8 time points per study |
| Selectivity index |
Ratio of DC50 between off-target and on-target proteins |
Global proteomics (TMT/DIA) across dose range, target vs off-target quantification |
One per degrader (8,000+ proteins) |
| Ubiquitination enrichment score |
Fold-change in ubiquitinated peptide abundance vs total protein level |
diGly enrichment + global proteomics normalization |
One per study condition |
Integrated Proteomics Platform for Targeted Protein Degrader Characterization
Comprehensive degrader characterization requires coordinated deployment of multiple proteomic strategies, each addressing specific dimensions of the degradation mechanism. Our platform integrates established best practices for global proteomic profiling, targeted quantification, ubiquitin remnant mapping, and interactome analysis within a unified quality-controlled workflow.
Global Proteomic Profiling — DIA and TMT Quantification
For unbiased proteome-wide assessment of degrader effects, cells or tissues are treated with the degrader molecule at specified concentrations and time points, lysed in urea-based buffer with protease and phosphatase inhibitors, and digested with trypsin/Lys-C using the filter-aided sample preparation (FASP) or S-Trap method. Peptides are analyzed by data-independent acquisition (DIA) on Orbitrap Astral or Q-TOF platforms for deep proteome coverage, or labeled with TMTpro reagents for multiplexed quantification across up to 18 conditions simultaneously. Raw data are processed using library-based or library-free DIA workflows (DIA-NN, Spectronaut) with false discovery rate control at both peptide and protein levels (FDR ≤ 1%). Differentially depleted proteins are identified by fold-change and statistical significance (limma, t-test with permutation-based FDR correction), with on-target degradation confirmed by comparison to vehicle-treated controls and known positive controls. For SILAC-based kinetic studies, our SILAC Degrader Kinetics service provides metabolic labeling-based degradation rate quantification with pulse-chase experimental designs.
Targeted MRM/PRM Quantification for DC50 Determination
For precise, routine quantification of target protein degradation across dose-response and time-course experiments, we deploy targeted MRM (on QQQ platforms) and PRM (on Orbitrap platforms) assays with isotope-labeled peptide standards (AQUA peptides or PSAQ proteins). Assays are developed for the target protein (3–5 proteotypic peptides) and a panel of potential off-targets identified from global discovery experiments, with heavy isotope-labeled internal standards for each target. Scheduled MRM acquisition with optimized collision energies and retention time windows delivers limits of quantification in the attomole range, enabling robust DC50, DC90, and Dmax calculation from 8–10 point dose-response curves with triplicate measurements per concentration.
Ubiquitin Remnant (diGly) Proteomics
To directly monitor degrader-induced ubiquitination — the mechanistic prerequisite for proteasomal degradation — we perform anti-diGly (K-ε-GG) antibody-based enrichment of ubiquitin remnant peptides from trypsin-digested proteomes. After tryptic digestion, peptides containing the diGly remnant (Gly-Gly, +114.043 Da) on ubiquitinated lysine residues are enriched using monoclonal antibody conjugates (PTMScan, K-ε-GG), and analyzed by LC-MS/MS with optimized gradient and acquisition parameters. Ubiquitinated peptides are identified by database searching with diGly as a variable modification, and quantified by label-free or TMT-based approaches. Comparison of diGly peptide abundance between degrader-treated and control conditions identifies degrader-induced ubiquitination events on the target protein and potential neo-substrates, providing direct mechanistic evidence for E3 ligase-dependent degradation. Our Ubiquitination Proteomics service provides dedicated diGly enrichment and quantification workflows optimized for degrader mechanism-of-action studies.
AP-MS Interactome Analysis for Ternary Complex Validation
Affinity purification-mass spectrometry (AP-MS) is deployed to validate the formation of the target-PROTAC-E3 ligase ternary complex — the critical event required for productive degradation. The target protein or E3 ligase is affinity-purified from degrader-treated cells using epitope tagging or specific antibodies, and co-purifying proteins are identified by LC-MS/MS with label-free quantification. Spectral counting and intensity-based quantification (iBAQ, MS1 filtering) are used to distinguish specific interactors from background contaminants, with statistical confidence assessed by SAINT or MiST scoring. Time-resolved AP-MS experiments capture the dynamic assembly of the ternary complex and subsequent ubiquitination machinery recruitment, providing mechanistic insights into the degradation event timeline.
Quality Control and Data Integration
Each proteomic module includes batch-specific quality control metrics: system suitability standards, calibration curves for targeted assays, inter-batch replicate correlation analysis (Pearson R ≥ 0.9 for global proteomics), and independent QC samples at defined degradation levels. For integrated multi-modal studies, data are normalized and cross-correlated across modules to ensure consistent quantification of degradation effects, with unified reporting that connects global discovery findings to targeted validation and mechanistic ubiquitinomics data.
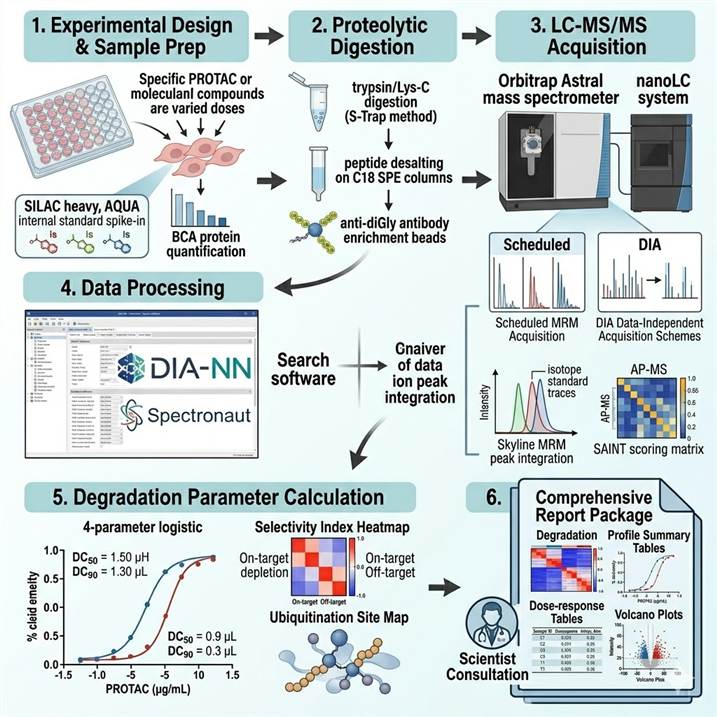
Modified Protein Degrader Proteomics Workflow: From Compound to Comprehensive Degradation Profile
Step 1: Experimental Design and Sample Preparation
Degrader dose-response and time-course treatment in relevant cell lines or in vivo models. Sample lysis in denaturing buffer with protease/phosphatase inhibitors, protein quantification (BCA assay), and quality control by SDS-PAGE. Internal standards (SILAC heavy spike-in, AQUA peptides) added at lysis for quantification accuracy.
Step 2: Proteolytic Digestion and Peptide Preparation
Trypsin/Lys-C digestion using S-Trap or FASP methods for global proteomics and diGly analysis. Peptide quantification, desalting by C18 SPE, and TMT labeling (if multiplexed). Anti-diGly antibody enrichment for ubiquitin remnant profiling. Affinity purification step for AP-MS interactome analysis.
Step 3: LC-MS/MS Data Acquisition
Global proteomics by DIA (Orbitrap Astral 120,000 resolution) or TMT (Orbitrap Eclipse SPS-MS3). Targeted MRM on QQQ (6500+) with scheduled acquisition. diGly-enriched peptides analyzed by HRAM LC-MS/MS with optimized gradient. AP-MS samples analyzed by label-free LC-MS/MS with ion mobility separation.
Step 4: Data Processing and Protein Identification
DIA data processed by DIA-NN or Spectronaut with library-based search. TMT data processed by Proteome Discoverer with CHIMERYS or Sequest HT. Targeted MRM data analyzed by Skyline or OS-Q. diGly data searched with MaxQuant or Proteome Discoverer with GlyGly modification. AP-MS data scored by SAINT or MiST.
Step 5: Degradation Parameter Calculation
On-target depletion quantified as % remaining vs vehicle control. DC50/DC90 calculated by 4-parameter logistic regression. Selectivity index computed from off-target DC50 ratios. Ubiquitination enrichment scored as diGly fold-change normalized to protein level. Degradation half-life calculated from time-course data.
Step 6: Integrated Reporting and Biological Interpretation
Comprehensive report with degradation profile summary, dose-response curves and DC50 tables, proteome-wide selectivity volcano plots, ubiquitination site maps, ternary complex validation data, pathway enrichment analysis of downstream effects, and a scientist consultation session for results interpretation in the TPD program context.
Protein Degrader Proteomics in Drug Discovery and Development
Proteomic characterization of targeted protein degraders spans the entire drug discovery pipeline — from early hit identification and mechanism-of-action validation through lead optimization, selectivity assessment, and preclinical candidate characterization. Our platform is configured to support the specific requirements of each application stage, from discovery-level global profiling to regulatory-grade targeted quantification.
PROTAC Discovery and Hit Validation
Early-stage PROTAC programs require rapid assessment of whether novel bifunctional molecules induce productive degradation of the intended target. Global proteomic profiling (DIA/TMT) of degrader-treated cells at a screening dose (e.g., 1 µM, 24 h) provides a comprehensive degradation footprint that confirms on-target depletion, reveals off-target effects, and identifies potential neo-substrates — all from a single experiment. This proteome-wide view enables early go/no-go decisions based on selectivity profiles, before committing resources to extensive medicinal chemistry optimization. For broader context on how PTM analysis integrates with drug discovery workflows, our PTMs in Drug Discovery and Development resource provides comprehensive guidance on applying PTM proteomics across the drug development pipeline.
Lead Optimization and Selectivity Assessment
As PROTAC leads progress through medicinal chemistry optimization, iterative proteomic profiling provides quantitative selectivity data that guides compound design. Dose-response global proteomics across multiple concentrations (e.g., 1 nM–10 µM) generates selectivity fingerprints that distinguish highly specific degraders from those with broad off-target effects. Parallel ubiquitinomics profiling confirms that target depletion proceeds through the intended E3 ligase mechanism, while AP-MS analysis validates ternary complex formation for each new chemical series. Our AP-MS Interactome Analysis service provides dedicated ternary complex validation for PROTAC and molecular glue development programs.
Molecular Glue Characterization
Molecular glues — small molecules that stabilize neo-protein-protein interactions between a target and an E3 ligase — present distinct characterization challenges compared to bifunctional PROTACs. Unlike PROTACs with defined target and E3 ligands, molecular glues require unbiased proteomic approaches to identify both the target and the E3 ligase involved in the degradation event. Our integrated degrader proteomics platform deploys AP-MS interactome analysis to identify glue-dependent neo-interactions, global proteomics to define the degradation profile, and ubiquitinomics to confirm mechanism-of-action — providing comprehensive characterization without requiring prior knowledge of the target-E3 ligase pair.
Resistance Mechanism Investigation
Acquired resistance to targeted protein degraders — through E3 ligase mutation or loss, target protein mutation, or adaptive proteostatic responses — is a recognized liability in TPD drug development. Our proteomics platform enables systematic investigation of resistance mechanisms through comparative proteomic profiling of sensitive versus resistant cell lines, ubiquitinomics to assess E3 ligase functionality, and global proteomics to identify compensatory pathways activated in response to chronic degrader treatment. This integrated approach has been instrumental in characterizing resistance to CRBN- and VHL-based PROTACs and developing next-generation degraders that overcome resistance through alternative E3 ligase recruitment.
In Vivo Degrader Pharmacodynamics
Translating in vitro degradation potency to in vivo efficacy requires robust pharmacodynamic readouts from tumor tissues, plasma, or surrogate matrices. Our targeted MRM/PRM assays, developed and validated against the target protein, provide the sensitivity and throughput needed for in vivo PD studies, with quantification limits compatible with limited tissue quantities from mouse xenograft models or patient biopsy samples. Dose- and time-dependent target depletion can be quantified alongside downstream pharmacodynamic biomarkers to establish the PK/PD relationship that guides clinical dose selection.
Case Study: DCAF1-Based PROTACs Overcome Intrinsic and Acquired Degrader Resistance
A 2024 study by Schröder et al. published in Nature Communications applied integrated proteomic analysis to characterize a novel class of DCAF1-based PROTACs that overcome resistance to CRBN- and VHL-recruiting degraders. This work demonstrates the power of proteomics-based degrader characterization for identifying resistance mechanisms and guiding next-generation TPD design.
Background: Most clinically advanced PROTACs recruit either CRBN or VHL as the E3 ubiquitin ligase. However, acquired resistance through CRBN mutation, loss of CRBN expression, or VHL pathway adaptation has been reported, limiting the long-term efficacy of first-generation PROTACs. A broadly expressed, essential E3 ligase with distinct substrate specificity — DCAF1 (DDB1- and CUL4-associated factor 1) — was identified as a potential alternative for PROTAC development, but whether DCAF1-based PROTACs could overcome CRBN/VHL resistance remained unknown.
Approach: The team developed selective, non-covalent DCAF1 ligands through structure-based design and functionalized them into DCAF1-based PROTACs targeting BRD9 (DBr-1) and BTK (DBt-10). Comprehensive proteomic characterization was performed using multiple complementary approaches: global TMT-based proteomics to assess degradation selectivity across the proteome, label-free quantitative proteomics for dose-response degradation profiling, chemoproteomics pull-down experiments to confirm DCAF1 binding specificity, and targeted MS-based quantification of known CRBN neo-substrates to evaluate off-target effects. Degradation profiles were compared directly between DCAF1-based PROTACs and their CRBN/VHL-based counterparts in both parental and degrader-resistant cell lines.
Key Findings:
- DCAF1-based PROTACs achieved potent and selective degradation of BRD9 (DC50 ~10 nM) and BTK (DC50 ~1 nM), with degradation profiles comparable to or exceeding CRBN/VHL-based PROTACs targeting the same proteins
- Global proteomics revealed that DCAF1-BRD9 PROTAC (DBr-1) showed high selectivity with minimal off-target depletion, distinct from the broader degradation footprint observed with CRBN-based BRD9 degraders that also depleted known CRBN neo-substrates (GSPT1, IKZF1/3)
- The DCAF1-BTK PROTAC (DBt-10) fully degraded BTK in cells with acquired resistance to CRBN-based PROTACs, demonstrating that DCAF1 recruitment overcomes CRBN-dependent resistance mechanisms
- DCAF1-BRD9 PROTAC retained full activity in cell lines with intrinsic VHL resistance, establishing DCAF1 as a viable E3 ligase for degrader programs where VHL-based approaches fail
- Chemoproteomics and ubiquitinomics confirmed that degradation proceeded through DCAF1-CRL4-dependent ubiquitination, with mechanistic validation through genetic rescue experiments using DCAF1 knockout and dominant-negative mutants
Significance: This study establishes DCAF1 as a broadly applicable E3 ligase for PROTAC development that overcomes both intrinsic and acquired resistance to CRBN- and VHL-based degraders. The comprehensive proteomic characterization — integrating global proteomics, chemoproteomics, ubiquitinomics, and targeted quantification — provided the multi-dimensional evidence required to validate the novel E3 ligase platform and guide resistance-overcoming degrader design. For researchers developing next-generation PROTACs and molecular glues, this work exemplifies the essential role of integrated proteomics in TPD drug discovery.
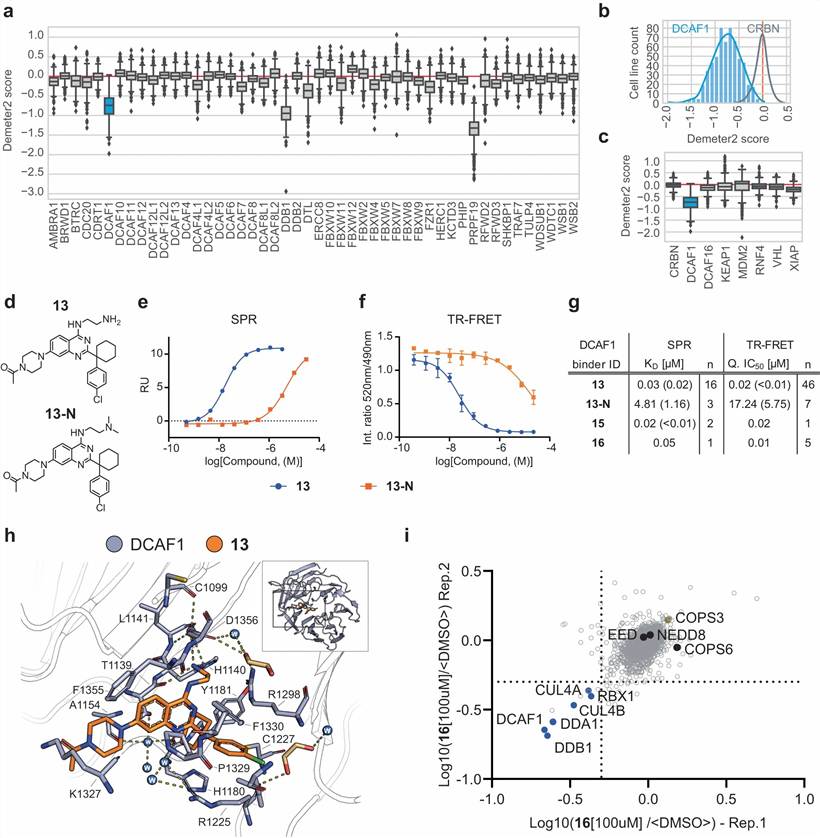
Figure 1 from Schröder et al. (2024). Proteomic characterization of DCAF1-based PROTACs overcoming degrader resistance. (a) Global TMT proteomics heatmap comparing degradation profiles of DCAF1, CRBN, and VHL-based PROTACs. (b) Dose-response degradation curves for DBr-1 (BRD9) and DBt-10 (BTK) with DC50 values. (c) BTK degradation by DBt-10 in CRBN-resistant cell lines. (d) Chemoproteomics pull-down confirming DCAF1 binder selectivity. (e) DCAF1-CRL4 ubiquitination model and genetic rescue validation. (CC BY 4.0)
Representative Protein Degrader Proteomics Data Outputs
Our modified protein degrader proteomics pipeline delivers multi-dimensional data outputs that provide a complete picture of degrader activity, selectivity, and mechanism. Below are representative examples of the key data types included in every project deliverable.
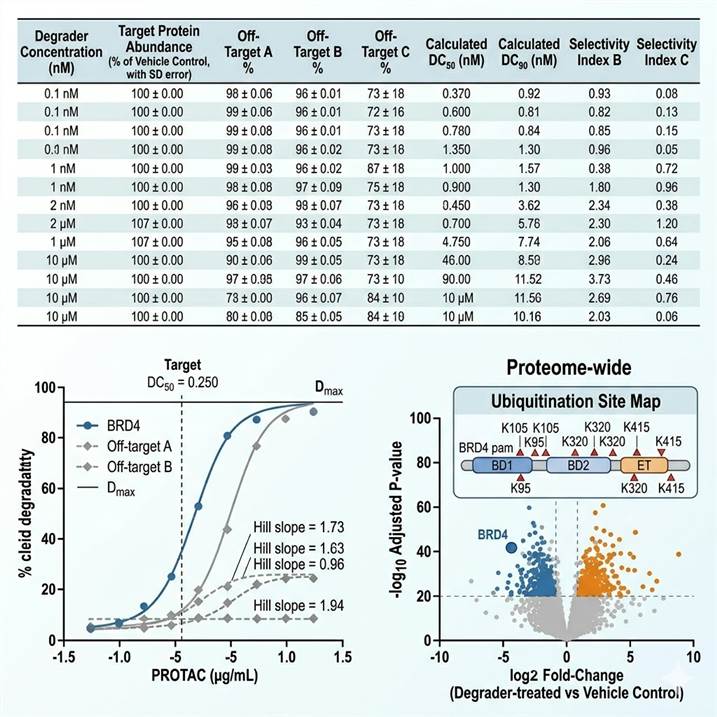
Representative protein degrader proteomics data. (Left) Degradation parameter table with degrader concentration (nM), target protein abundance (% of vehicle control), key off-target protein abundance, calculated DC50/DC90 values (nM), and selectivity index for each off-target. (Center) Dose-response degradation curves with 4-parameter logistic regression — target protein (blue) and off-target proteins (gray) — showing DC50, Dmax, and Hill slope values. (Right) Proteome-wide selectivity visualization combining a volcano plot (log2 fold-change vs -log10 p-value) with annotated target and off-target proteins, alongside a ubiquitination site map showing degrader-induced diGly peptides on the target protein with assigned ubiquitination site positions.
Every data deliverable includes raw LC-MS/MS files, processed quantification tables with quality control metrics, interactive visualization files for dose-response curves and selectivity profiling, ubiquitination site annotation with sequence coverage maps, and a scientist consultation session for results interpretation within the context of the TPD program. Custom reporting formats aligned with regulatory submission requirements (including ICH M10-compliant bioanalytical documentation for targeted MRM assays) are available on request.
Why Choose Our Modified Protein Degrader Proteomics Services
Multi-Modal Integrative Platform
Our degrader proteomics platform uniquely integrates four complementary analytical modalities — global proteomics (DIA/TMT), targeted MRM quantification, ubiquitin remnant diGly profiling, and AP-MS interactome analysis — within a unified workflow. This integrative approach ensures that degradation selectivity, potency, mechanism, and target engagement are characterized simultaneously from the same experimental system, eliminating the cross-study variability inherent in piecemeal characterization strategies.
TPD-Specific Quantification Expertise
Our team has deep expertise in the specific analytical challenges of targeted protein degradation, including accurate DC50 determination from proteomics data, ubiquitination site mapping for mechanism-of-action validation, neo-substrate identification, and E3 ligase selectivity profiling. We have supported PROTAC and molecular glue programs across multiple therapeutic areas, from early discovery through preclinical candidate selection.
Cross-Platform Validation and Regulatory Readiness
All targeted degradation findings from global discovery proteomics are cross-validated by independent MRM/PRM assays with isotope-labeled internal standards, ensuring that DC50, selectivity, and degradation kinetics data meet the analytical rigor expected for regulatory submissions. Our targeted quantification workflows are developed with ICH M10 bioanalytical method validation principles, supporting the transition from discovery to regulated bioanalysis.
Integrated Drug Discovery Ecosystem
Our degrader proteomics service operates within a comprehensive PTM and drug discovery platform that includes covalent drug target engagement profiling, kinase activity assays, ubiquitin enzyme assays, and epigenetic PTM characterization. This ecosystem enables seamless integration of degradation data with broader drug discovery workflows, from target identification through mechanism-of-action studies to preclinical biomarker development.
Related Services
Our modified protein degrader proteomics service is supported by a broader drug discovery and PTM characterization platform offering complementary analytical capabilities for targeted protein degradation programs.
- Covalent Drug PTM Profiling — Chemoproteomic profiling of covalent inhibitor target engagement and off-target reactivity for covalent drug discovery programs
- Reactive Cysteine Target Engagement Assay — LC-MS-based assessment of covalent probe binding to reactive cysteine residues for target engagement and selectivity profiling
- DUB and Ubiquitin Enzyme Activity Assays — Activity-based profiling of deubiquitinating enzymes and ubiquitin conjugation machinery for ubiquitin pathway drug discovery
- HDAC/HAT Activity Assays — Targeted activity measurements for histone deacetylase and acetyltransferase enzymes in epigenetic drug discovery
- Kinase Activity Profiling — Comprehensive kinase activity profiling for drug selectivity assessment and mechanism-of-action studies
- Epigenetic PTM Research — Specialized PTM analysis services for epigenetic research, including histone modifications and chromatin-associated protein characterization
- Global PTM Profiling — Broad multi-PTM discovery analysis across diverse protein modification classes for integrated drug discovery studies
- MS-Based PTM Analysis — Comprehensive mass spectrometry platform for protein-level PTM discovery, quantification, and characterization
- Bottom-Up MS-Based PTM Analysis — Deep PTM discovery and quantitative profiling using the shotgun proteomics approach
- Open-Search PTM Discovery — Unbiased open-search approach for detecting unexpected modifications across proteomics datasets
Frequently Asked Questions
What is the difference between DC50 and IC50 in the context of targeted protein degradation?
DC50 (degradation concentration 50%) measures the degrader concentration required to achieve 50% depletion of the target protein, quantified by proteomics. IC50 (inhibitory concentration 50%) measures the concentration required for 50% inhibition of a biochemical or cellular activity. For degraders, the DC50 is often lower than the IC50 because degradation is catalytic — a single degrader molecule can induce degradation of multiple target protein molecules — whereas inhibition typically requires stoichiometric occupancy. Both parameters are valuable: DC50 defines degradation potency, while IC50 (or pIC50 from functional assays) defines residual activity of the non-degraded fraction.
How do you distinguish on-target degradation from off-target effects in proteomics data?
On-target degradation is confirmed through multiple orthogonal criteria: the target protein must show statistically significant depletion (adjusted p-value < 0.05, fold-change below threshold) that is dose-dependent and time-dependent; depletion must be rescued by E3 ligase knockout or proteasome inhibition; the target should not be depleted by a negative control compound (inactive enantiomer or null PROTAC); and the target mRNA level should remain unchanged (confirmed by RT-qPCR or RNA-seq). Off-targets are defined as proteins showing significant depletion that do not meet these criteria and are typically fewer in number and depleted with lower potency (higher DC50) than the intended target.
What sample types are compatible with your degrader proteomics service?
Our service is compatible with a wide range of sample types. For in vitro studies, we routinely process adherent and suspension cell lines, PBMCs, and primary cells. For in vivo studies, we accept fresh-frozen or cryopreserved tissues (tumor xenografts, organ tissues, plasma/serum). Typical sample requirements: cell pellets ≥5 × 10⁶ cells per condition for global proteomics (≥1 × 10⁷ for ubiquitinomics), tissue ≥10 mg (wet weight) for global proteomics, and ≥50 µL plasma for targeted MRM assays. We recommend triplicate biological replicates for statistically robust degradation profiling.
What is the typical timeline and throughput for degrader proteomics studies?
Timelines vary by study scope. A standard degrader dose-response study (single target, 8 concentrations, triplicates, global proteomics + MRM validation) typically requires 4–6 weeks from sample receipt to final report. A comprehensive multi-modal characterization (global proteomics + ubiquitinomics + AP-MS across multiple time points and concentrations) requires 8–12 weeks. For targeted MRM-only assays (DC50 determination for established targets), we deliver results within 2–3 weeks. Throughput ranges from single compound deep characterization to panel screening of 10–20 degraders in parallel using TMT multiplexing.
Can you characterize molecular glues that do not have a pre-defined target or E3 ligase?
Yes — our integrated degrader proteomics platform is specifically designed to characterize molecular glues where neither the target nor the E3 ligase is known a priori. Our approach combines: (1) global proteomic profiling to identify all depleted proteins (potential targets) and degraded neo-substrates; (2) AP-MS interactome analysis with unbiased bait identification to discover glue-dependent protein-protein interactions; (3) chemoproteomics with immobilized glue derivatives to identify direct binding proteins; and (4) ubiquitinomics to confirm degradation mechanism. This multi-pronged strategy provides comprehensive characterization without requiring prior knowledge of the molecular glue's target or mechanism.
How do you validate that target degradation is proteasome-dependent and E3 ligase-specific?
Mechanistic validation of proteasome-dependent degradation uses multiple complementary approaches: (1) pre-treatment with proteasome inhibitors (MG132, bortezomib, carfilzomib) should rescue target protein levels; (2) E3 ligase knockout or knockdown (CRISPR or siRNA) should abolish or reduce degradation; (3) NEDD8-activating enzyme inhibitor MLN4924 treatment should block Cullin-RING ligase-dependent degradation; (4) ubiquitinomics (diGly enrichment) should show increased ubiquitination of the target upon degrader treatment; and (5) for PROTACs, an inactive control compound (lacking either the target or E3 ligand) should show no degradation. We routinely incorporate these validation experiments as part of comprehensive degrader characterization studies.
What bioinformatics and statistical methods are used for degrader proteomics data analysis?
Our bioinformatics pipeline includes multiple analysis modules tailored to TPD data interpretation: differential protein abundance analysis using limma or DEP with FDR correction; dose-response curve fitting with 4-parameter logistic regression for DC50/DC90 calculation; selectivity scoring with ranked off-target lists and selectivity index calculation; ubiquitination site annotation with mapping to domain structures and functional sites; pathway enrichment analysis (GO, KEGG, Reactome) for downstream effect deconvolution; and multi-omics data integration for combined global proteomics + ubiquitinomics + AP-MS studies. Data are reported with interactive visualization dashboards for exploratory analysis.
References
- Schröder M, Renatus M, Liang X, Meili F, Zoller T, Ferrand S, Gauter F, Li X, Sigoillot F, Gleim S, Stachyra TM, Thomas JR, Begue D, Khoshouei M, Lefeuvre P, Andraos-Rey R, Chung B, Ma R, Pinch B, Hofmann A, Schirle M, Schmiedeberg N, Imbach P, Gorses D, Calkins K, Bauer-Probst B, Maschlej M, Niederst M, Maher R, Henault M, Alford J, Ahrne E, Tordella L, Hollingworth G, Thomä NH, Vulpetti A, Radimerski T, Holzer P, Carbonneau S, Thoma CR. DCAF1-based PROTACs with activity against clinically validated targets overcoming intrinsic- and acquired-degrader resistance. Nat Commun. 2024;15:275.
- Wang C, Zhang Y, Chen W, Wu Y, Xing D. New-generation advanced PROTACs as potential therapeutic agents in cancer therapy. Mol Cancer. 2024;23:110.
- Wang S, He F, Tian C, Sun A. From PROTAC to TPD: advances and opportunities in targeted protein degradation. Pharmaceuticals. 2024;17(1):100.
For research use only. Not for use in diagnostic procedures.