Why LC-MS/MS for Histone PTM Analysis — Beyond Antibody-Dependent Approaches
Chromatin immunoprecipitation (ChIP)-based methods have dominated histone modification research for two decades, but they carry inherent limitations: each antibody detects only a single mark, antibody specificity is variable across histone sequence contexts, and ChIP cannot distinguish combinatorial marks on the same histone tail. These constraints become critical when studying the 50+ known histone PTMs and their thousands of potential combinatorial configurations.
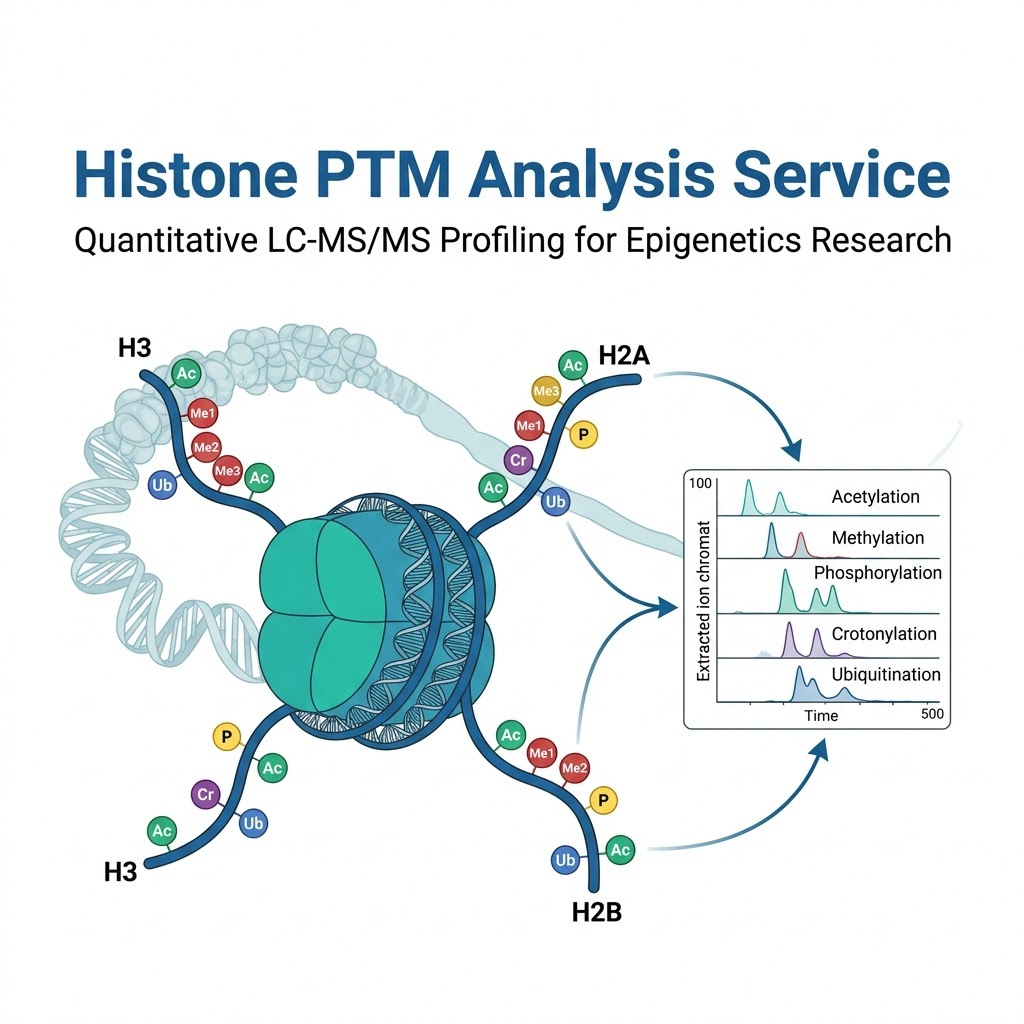
LC-MS/MS-based histone PTM analysis overcomes these limitations entirely. In a single analytical run, our platform simultaneously identifies and quantifies acetylation, methylation (mono-, di-, tri-), phosphorylation, crotonylation, ubiquitination, and other modifications across histone H3, H4, H2A, H2B, and linker histone H1. Site-level localization is determined by diagnostic fragment ions — not by antibody availability — and relative quantification is achieved by extracted ion chromatogram integration with CVs consistently below 15%. The result is an unbiased, quantitative map of the histone PTM landscape that ChIP-based methods cannot match.
Histone Marks We Profile — Acetylation, Methylation, Phosphorylation, Crotonylation & Ubiquitination
Our standard histone PTM panel covers the most biologically relevant marks across all core histones. Custom panels can be configured to prioritize specific modification types or histone variants for project-specific needs.
Acetylation
Lysine acetylation (Kac) neutralizes the positive charge on histone tails, reducing DNA-histone interaction and promoting open chromatin. Key marks include H3K9ac, H3K14ac, H3K27ac (active enhancers), H4K5ac, H4K8ac, H4K12ac, and H4K16ac. Our workflow quantifies acetylation stoichiometry at each site, enabling correlation with transcriptional output. For broader acetylome profiling beyond histones, our Acetylomics Analysis service provides complementary proteome-wide coverage.
Methylation
Lysine and arginine methylation are among the most information-rich histone marks, with distinct methylation states (me1/me2/me3) carrying different functional consequences. We resolve all three methylation states at key residues including H3K4 (active promoters), H3K9 (heterochromatin), H3K27 (Polycomb repression), H3K36 (transcriptional elongation), H3K79, and H4K20. Arginine symmetric and asymmetric methylation (H3R2, H3R8, H3R17, H4R3) are also quantified. For proteome-wide methylation analysis, our Methylation Proteomics service extends beyond histones to the full methyl-proteome.
Phosphorylation
Histone phosphorylation is tightly linked to chromosome condensation during mitosis (H3S10ph, H3S28ph), DNA damage signaling (H2AX S139ph — γH2AX), and transcriptional regulation. Our panel quantitatively monitors these dynamic marks across cell cycle stages or in response to DNA damage.
Crotonylation
The recently discovered lysine crotonylation (Kcr) is structurally and functionally distinct from acetylation, marking active promoters and potential enhancers with unique regulatory properties. Our assay includes crotonylation detection alongside the classical marks.
Ubiquitination
Histone ubiquitination (particularly H2BK120ub and H2AK119ub) plays essential roles in transcriptional elongation and Polycomb repression. Ubiquitinated histone peptides are identified by the characteristic GG remnant on modified lysine residues after trypsin digestion.
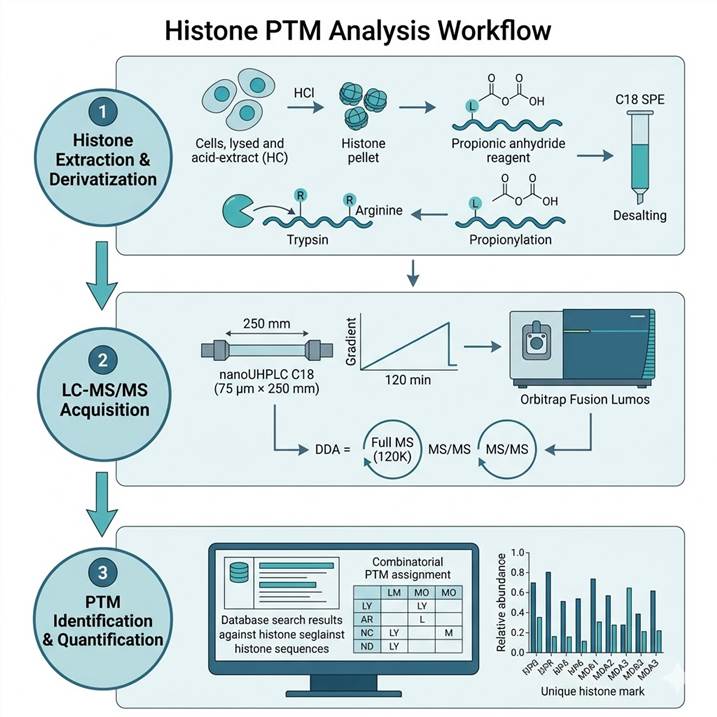
Histone PTM Analysis Workflow — From Sample to Site-Specific Quantification
Our histone PTM workflow incorporates chemical derivatization steps that are essential for chromatographic resolution of the highly basic, hydrophilic histone N-terminal tails, enabling detection of combinatorial modifications on single peptides.
Step 1: Histone Extraction & Derivatization
- Acid extraction of core histones from cell pellets or tissue
- Propionylation of lysine residues to block tryptic cleavage at K
- Trypsin digestion (cleaves only at R residues after propionylation)
- Second propionylation to cap newly exposed N-termini after digestion
- Desalting and concentration by C18 SPE
Step 2: LC-MS/MS Acquisition
- Reversed-phase C18 separation on nanoUHPLC (75 μm × 250 mm column)
- 120-minute gradient optimized for histone peptide resolution
- Orbitrap Fusion Lumos or Eclipse in DDA or PRM mode
- Full MS at 120,000 resolution; MS/MS in ion trap or Orbitrap
- Targeted PRM assays for low-abundance marks where needed
Step 3: PTM Identification & Quantification
- Database search against histone sequences with variable PTM modifications
- Combinatorial PTM assignment on multiply-modified peptides
- Relative quantification by XIC peak area for each modified form
- Normalization to total histone or unmodified reference peptide
- Results reported as relative abundance (%) for each PTM mark per site
For broader modification mapping across the entire proteome, our Global PTMs Profiling service and Pan PTM Proteomics provide complementary coverage across diverse PTM types.
Research Applications — Epigenetics Discovery to Drug Development
Histone PTM analysis supports a broad range of research programs, from basic chromatin biology to preclinical epigenetic drug development.
Epigenetic Drug Mechanism of Action Studies
HDAC inhibitors, HMT inhibitors, and demethylase inhibitors produce characteristic changes in histone PTM landscapes that serve as pharmacodynamic biomarkers. Our quantitative profiling maps these changes across dose and time, providing direct evidence of target engagement and off-target effects at the chromatin level.
Cancer Epigenetics and Biomarker Discovery
Global alterations in histone modification patterns are hallmarks of cancer. H3K27me3 loss in aggressive lymphomas, H3K9me3 alterations in AML, and global H4K16ac loss in multiple cancer types represent well-established epigenetic lesions. Our service enables systematic comparison of histone PTM profiles between tumor and normal samples, identifying candidate biomarkers and pathway-level epigenetic dysregulation.
Stem Cell Differentiation and Development
Histone PTM landscapes undergo coordinated reprogramming during embryonic stem cell differentiation and lineage commitment. Temporal profiling of bivalent domains (H3K4me3 + H3K27me3) and activation marks provides mechanistic insight into cell fate decisions and can guide directed differentiation protocols.
Sample Requirements for Histone PTM Analysis
Our histone PTM workflow is compatible with a range of sample types from cultured cells to tissue biopsies. The recommended input amounts below are based on the propionylation-based bottom-up workflow.
| Sample Type |
Recommended Amount |
Storage & Shipping |
Critical Notes |
| Cultured cells (mammalian) |
1 × 10⁶ – 1 × 10⁷ cells |
Snap-freeze pellet; −80 °C, ship on dry ice |
Minimize cell passage number for consistent histone modification profiles. Include HDAC inhibitor (e.g., 10 mM Na butyrate) in wash buffer if studying acetylation |
| Tissue biopsy |
5–50 mg tissue |
Snap-freeze in liquid nitrogen; −80 °C, ship on dry ice |
Homogenize in histone extraction buffer with protease inhibitors + HDAC inhibitors. Avoid formalin fixation |
| FACS-sorted cells |
5 × 10⁴ – 1 × 10⁶ cells |
Sorted directly into lysis buffer or snap-frozen pellet |
Our workflow can detect 60+ histone peptides from as few as 1,000 cells (see Case Study) |
| Histone-enriched fractions |
5–25 µg total histones |
−80 °C, ship on dry ice |
Provide histone purity estimate (SDS-PAGE or BCA). Acid-extracted histones preferred |
Deliverables — Publication-Ready Histone PTM Reports
Every histone PTM project delivers a complete data package designed for direct use in manuscripts, grant applications, and regulatory submissions.
Standard Deliverables
- Histone PTM identification table — every modified peptide with mark type, site localization, combinatorial modification assignments, and MS/MS spectral annotation
- Relative quantification results — percent abundance for each PTM mark at each site, normalized to total histone or reference peptide, with intra-run CVs
- Comparative PTM heatmaps — cross-condition mark landscape visualization for rapid pattern recognition
- Extracted ion chromatograms — overlaid traces for modified and unmodified peptide pairs
- Method appendix — detailed protocol suitable for manuscript methods and regulatory submission
- Interpretive project summary — contextual overview highlighting significant mark changes and biological implications
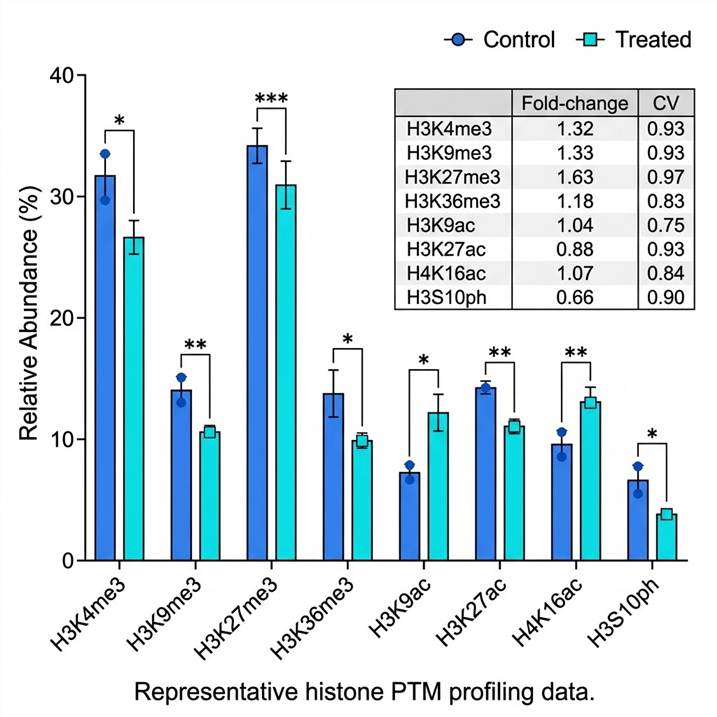
Case Study: Targeted Histone PTM Detection from 1,000 Cells — From Method Optimization to Clinical Application
To demonstrate the sensitivity and reproducibility of LC-MS-based histone PTM analysis, we highlight a study by Abshiru et al. (PLOS ONE, 2020) that developed and validated a targeted LC-MS/MS method for histone modification analysis from as few as 1,000 cells — representing a >100-fold improvement in sample input compared to standard histone PTM workflows.
Methodological Innovations
The study systematically optimized two critical sample preparation parameters. First, omitting the nuclear isolation step increased detected histone peptide peak areas by 2.6-fold on average, by eliminating sample losses during subcellular fractionation. Second, reducing the propionylation derivatization from two rounds to one increased peak areas by an additional 4.2-fold, by minimizing peptide loss during cleanup steps. These combined improvements delivered approximately 10-fold higher signal for the same starting cell number (Fig. 1–2, Abshiru et al.).
Sensitivity and Scalability
Using the optimized workflow, the authors demonstrated linear quantification across a 1,000-fold range of cell inputs (10³ to 10⁶ HeLa-S3 cells), with 89 histone peptides quantified from 10⁶ cells and 61 peptides from just 10³ cells (Fig. 3). The method was then applied to primary human cells: from only 1,000 normal bone marrow CD34+ hematopoietic stem/progenitor cells, 61 distinct histone peptide forms were detected and quantified, covering acetylation and methylation marks on H3 and H4 N-terminal tails (Fig. 4).
Clinical Application: AML Patient Samples
The clinical utility of the method was demonstrated by profiling histone modifications from 1,000 primary acute myeloid leukemia (AML) cells per patient. Across six AML patient samples (10⁶ cells each), the authors identified a striking dichotomy in H3K9me2 and H3K9me3 levels — with a subset of patients showing high H3K9me2/3 and others showing low levels. Notably, patients with elevated H3K9me3 also showed higher mRNA expression of SETDB1, the methyltransferase responsible for H3K9 trimethylation (Fig. 5). This finding suggests that histone PTM profiling can uncover underlying epigenetic regulatory mechanisms and potential biomarkers directly from limited clinical material.
Conclusion
This study establishes that targeted LC-MS/MS histone PTM analysis can deliver robust, quantitative data from sample amounts previously accessible only to antibody-based methods — but with the multiplexing capacity and site specificity that ChIP approaches cannot provide. The same analytical framework underlies our service, scaled and adapted for broader PTM panel coverage across diverse sample types.
Source
Abshiru, N. A.; Sikora, J. W.; Camarillo, J. M.; Morris, J. A.; Compton, P. D.; Lee, T.; Neelamraju, Y.; Haddox, S.; Sheridan, C.; Carroll, M.; Cripe, L. D.; Tallman, M. S.; Paietta, E. M.; Melnick, A. M.; Thomas, P. M.; Garrett-Bakelman, F. E.; Kelleher, N. L. PLOS ONE 2020, 15, e0240829. DOI: 10.1371/journal.pone.0240829. Figures 1–5 provide the complete method optimization and application data.
Histone PTM Analysis: Frequently Asked Questions
How many histone marks can you detect in a single experiment?
In a standard experiment using our propionylation-based bottom-up workflow, we detect and quantify 50–90 distinct histone peptide forms spanning acetylation, methylation (me1/me2/me3), and phosphorylation marks across histones H3 and H4. When extended to H2A, H2B, and H1, the total exceeds 120 modified peptides. The exact number depends on sample type, cell state, and starting amount.
What is the minimum cell number required for histone PTM analysis?
Our standard workflow recommends 1 × 10⁶ cells for comprehensive coverage. For sample-limited studies, we have demonstrated robust quantification of 60+ histone peptides from as few as 1,000 cells using targeted methods (see Case Study). Please contact our team to discuss feasibility for your specific sample type and research question.
Can you distinguish between different methylation states (me1 vs me2 vs me3)?
Yes. Monomethylation (+14 Da), dimethylation (+28 Da), and trimethylation (+42 Da) produce distinct, resolvable mass shifts that are unambiguously identified at the MS1 level and confirmed by MS/MS fragmentation. Our quantification reports each methylation state independently at each site.
How do you handle combinatorial PTMs on the same histone tail?
Combinatorial PTMs — multiple modifications on the same histone peptide — are identified by the combined mass shifts and confirmed by MS/MS fragmentation. Our bioinformatics pipeline assigns combinatorial marks and reports the relative abundance of each co-occurring modification pattern.
What types of histone variants can you analyze?
Our workflow covers canonical histones (H3.1, H3.2, H3.3, H4, H2A, H2B) and variant histones (H2A.Z, H2A.X, macroH2A, H3.Y, H3.5, CENP-A) when present in sufficient abundance. Histone variant-specific peptides are identified by sequence-unique tryptic peptides that distinguish variants from canonical forms.
References
- Abshiru, N. A.; Sikora, J. W.; Camarillo, J. M.; Morris, J. A.; Compton, P. D.; Lee, T.; Neelamraju, Y.; Haddox, S.; Sheridan, C.; Carroll, M.; Cripe, L. D.; Tallman, M. S.; Paietta, E. M.; Melnick, A. M.; Thomas, P. M.; Garrett-Bakelman, F. E.; Kelleher, N. L. "Targeted Detection and Quantitation of Histone Modifications from 1,000 Cells" PLOS ONE 2020, 15, e0240829.
- Sidoli, S.; Bhanu, N. V.; Karch, K. R.; Wang, X.; Garcia, B. A. "Complete Workflow for Analysis of Histone Post-Translational Modifications Using Bottom-Up Mass Spectrometry" Mol. Cell. Proteomics 2016, 15, 1144–1156.
- Huang, H.; Lin, S.; Garcia, B. A.; Zhao, Y. "Quantitative Proteomic Analysis of Histone Modifications" Cell 2015, 163, 1414–1426.

.jpg)

